


Roche SBX deep dive into the real specs
What is hidden behind the numbers presented by Roche?

Roche presented their SBX sequencing system via a webinar and at AGBT25, and I live tweeted on Bluesky about it.
Here is a deep dive into the details of the presentation, and why I think there are some hidden truths about the specs presented that are worth highlighting before everyone gets carried away about what this new technology means for Next-Generation Sequencing. The pictorial representation shown below is “too generic” to define what Roche SBX is as a product, so we better dive into it for the full details.

The webinar started by the big poncho at Roche outlining the contents of the presentation.

The presenter emphasized that Roche has a deep history of commitment to innovation. We know Roche is company making an annual revenue of $8B in diagnostics, across a multitude of product lines.
They do about 30 billion tests per year, have more than 100,000 employees worldwide including 3 Nobel prizes.
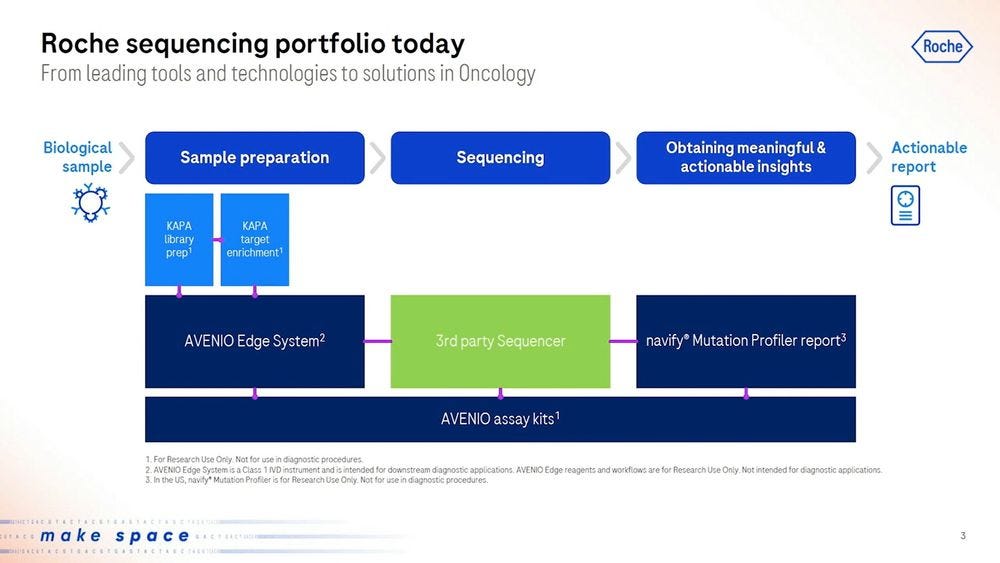
Going down to the specifics of diagnostics via sequencing, Roche has been selling their AVENIO Edge sample preparation products, including KAPA library prep and KAPA target enrichment, which would then be put into a 3rd party sequencer (aka an Illumina machine), to then be analyzed with Roche’s software like the navify Mutation Profiler for Actionable reports.
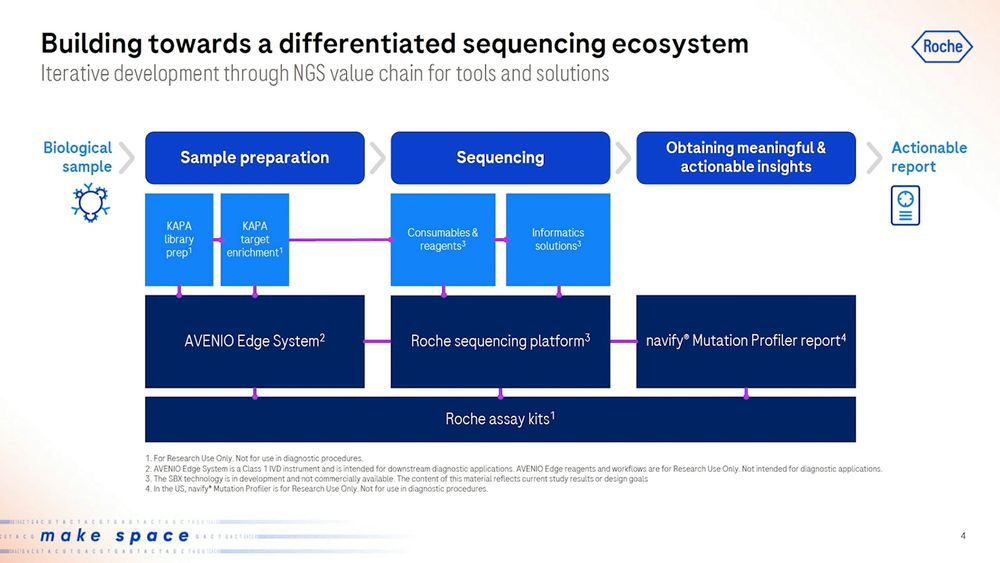
Roche presented the new Roche sequencing platform, with Consumables and reagents together with Informatics solutions that plug directly into the downstream software. This will be sold as a standalone RUO (Research Use Only) product to the likes of academics and researchers to use, but also as a clinical offering later on.

After outlining the strategy, the Head of SBX Technology, co-founder and CEO of Stratos Genomics (2007-2020) stepped forward to give us the juice on this new SBX sequencing technology.

The welcoming remarks by Mark, who has been in the field for the last 18 years, he did mention in several occasions that the methodology they were proposing took a long time to come along, and that many people had told them it wouldn’t work.
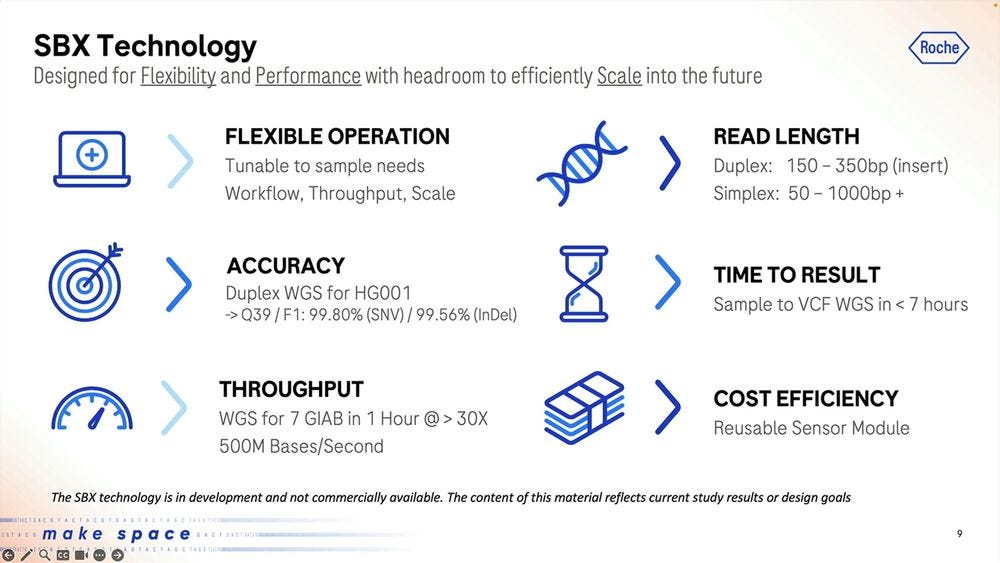
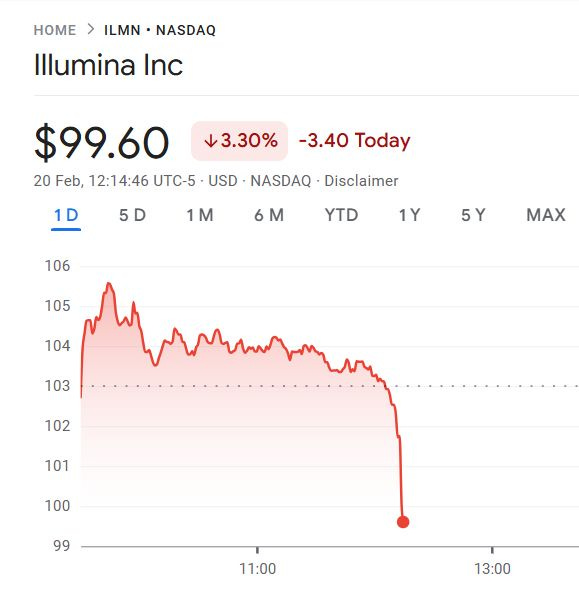
The main specs of the SBX Technology as of today are a short-read system that produces two types of reads: Duplex and Simplex, with a Duplex method running on the sequencer instrument for 4 hours producing about 15 billion single-end reads with an original insert of 150-350 base pairs.
So this is pretty much where Illumina is currently making 99% of their revenue, in short-read NGS of 2x150bp kits (300 cycles or 300 base pairs), hence the reason why the stock took a dive on the day Roche presented their SBX specs.
The offering of this Roche SBX technology is composed of:
One instrument for the SBX chemistry (library prep) and,
One instrument for the sequencing.
These may become a single instrument in the future if the customer sees benefit in combining them. We’ve seen companies like Element Bio innovate in integrating the whole process so that the user simply needs to inject the extracted DNA into the instrument, and library prep (whole genome or enrichment) and sequencing happen seamlessly from then onwards.

Roche did a beta trial of the SBX technology with two expert outlets in NGS: the Hartwig Hospital Foundation in Amsterdam and the Broad Institute in Boston.
The first step of the process in SBX is to convert the initial DNA molecule (or cDNA if it came from RNA) into an expandomer surrogate molecule, which is used to rescale the signal-to-noise ratio and make this nanopore-based detection method work in the Q20 range at the single-molecule single-event level. In comparison, Oxford Nanopore does this natively, and already achieves Q25+ single-molecule single-event accuracy, but it took them 10 years to reach this point. Copycat nanopore companies in China are at 10x lower accuracy level currently, with MGI Tech and others attempting to improve their technologies.
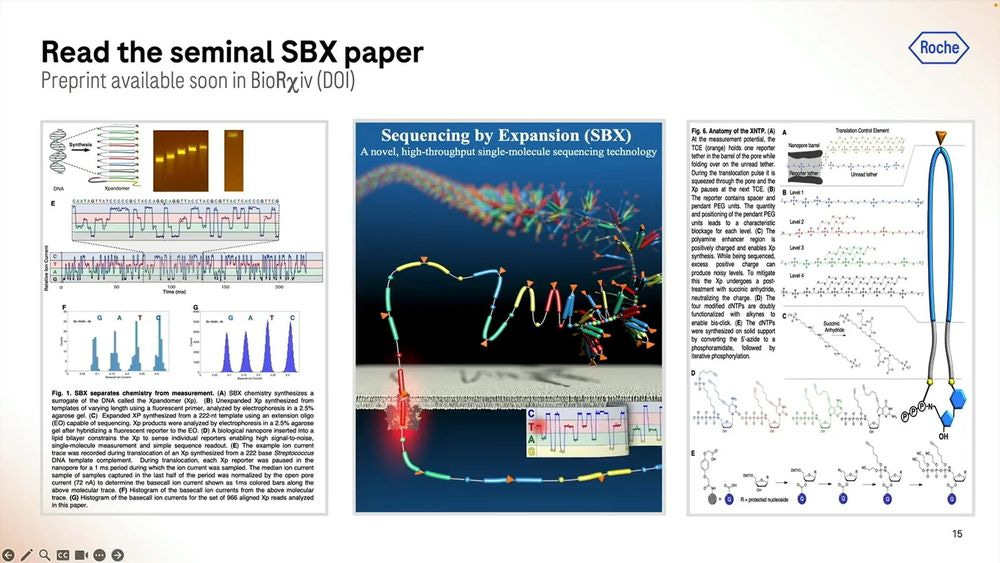
SBX is now available as a 60-70 page preprint for the Science geeks to “geek out” about the details of this interesting piece of biochemistry.
These expandomers are so big, they don’t look like DNA at all, and hence Roche/Stratos had to modify the enzymes (polymerases and others) to adapt to this type of new synthetic chemistry.
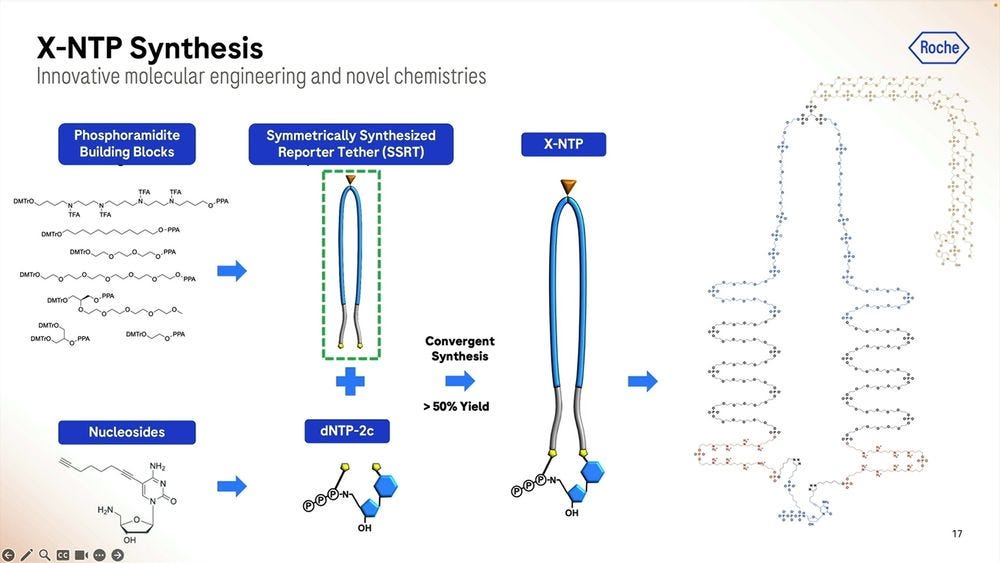
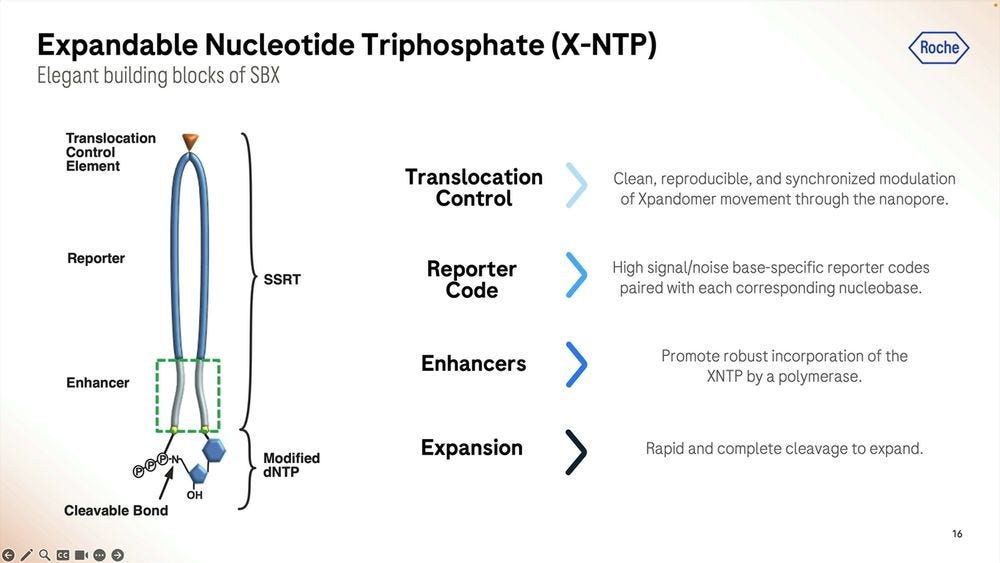
Roche/Stratos iterated over thousands of custom molecules, the XNTPs (units equivalent to nucleotides, or NTPs) and SSRTs (tethers) until they found the right approach. After optimising for many years, they now get a purified yield of over 50%, which for the (bio-)chemists reading this, they’ll be able to say that’s a very efficient process.
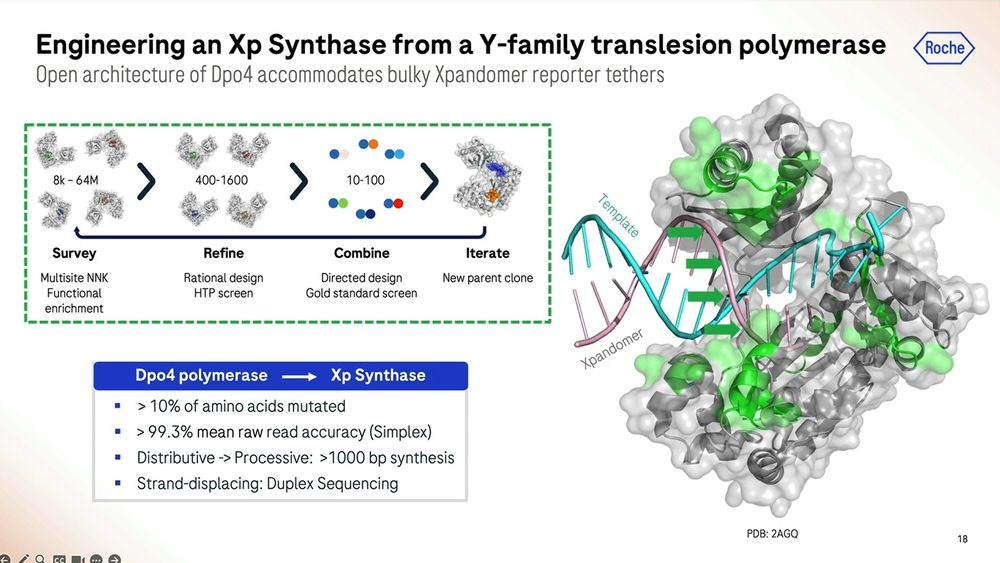
Roche/Stratos used a combination of rational design and high-throughput screening to iterate over thousands and thousands of polymerase versions. The molecule is so far away from what is seen in nature that it should really be renamed as an XP-synthase enzyme.
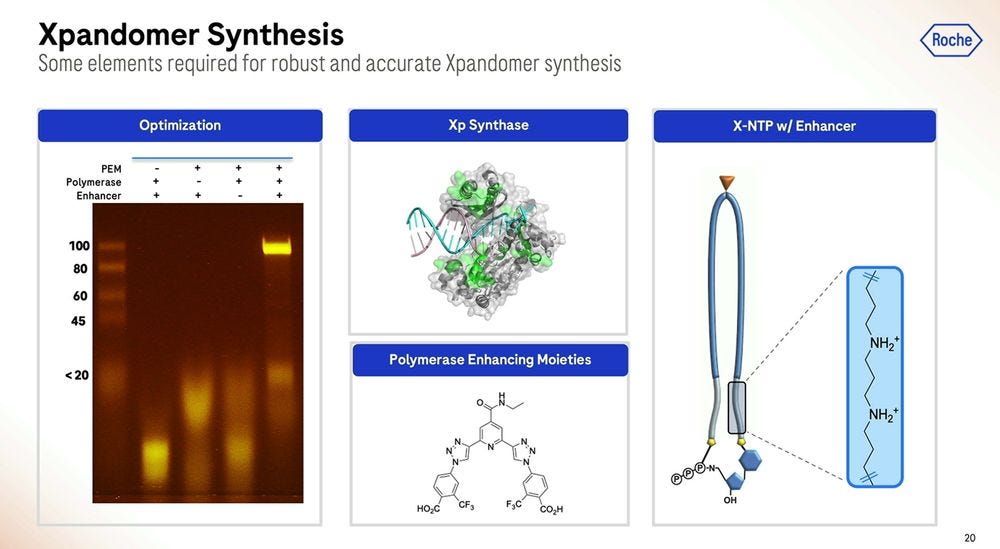
The polymerase enhancing moiety is the bit that glues the whole thing together, which was also crucial to get right. The gel on the left shows how much better (how much cleaner bands) the current version produces compared to previous iterations (smears of complex partial products).
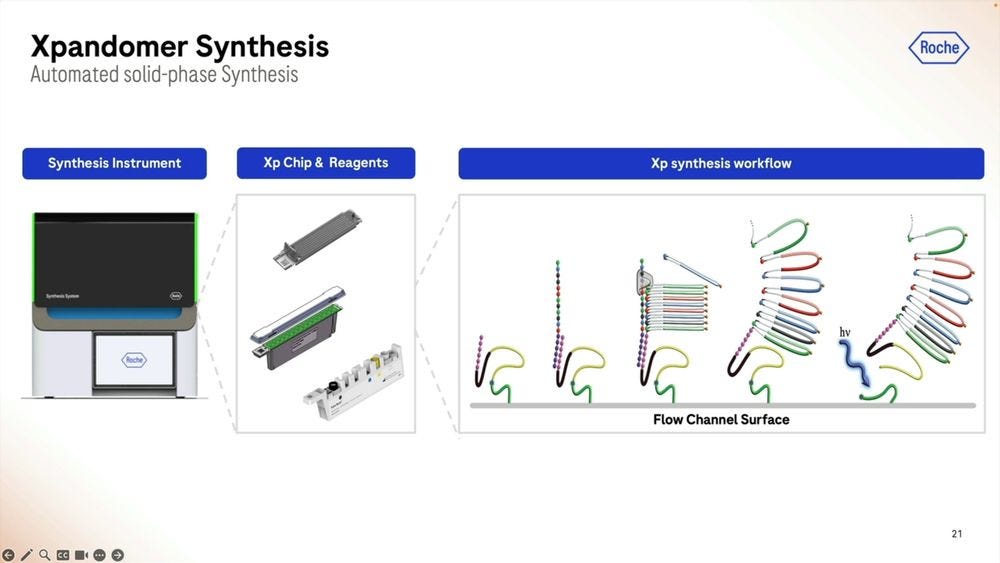
The Xp chip is what’s loaded with the sample that’s going to be library prepped, where the synthesis part takes place, the expandomers are then ready to go into the second instrument, the sequencer. The first instrument, the synthesizer, is the narrow one, compared to the sequencer, which is still a benchtop instrument, but wider, the width of an American fridge-freezer.

Since these two instruments couple the two step process, one can combine multiples of the first to match up the throughput of the second. In my debriefing conversation with James Hadfield at Astrazeneca, we discussed a ratio of 4 to 1 between the library prep instruments (4) and the sequencer (1), especially for people wanting to do rapid MRD applications (1 hour runs rather than 4 hour runs).
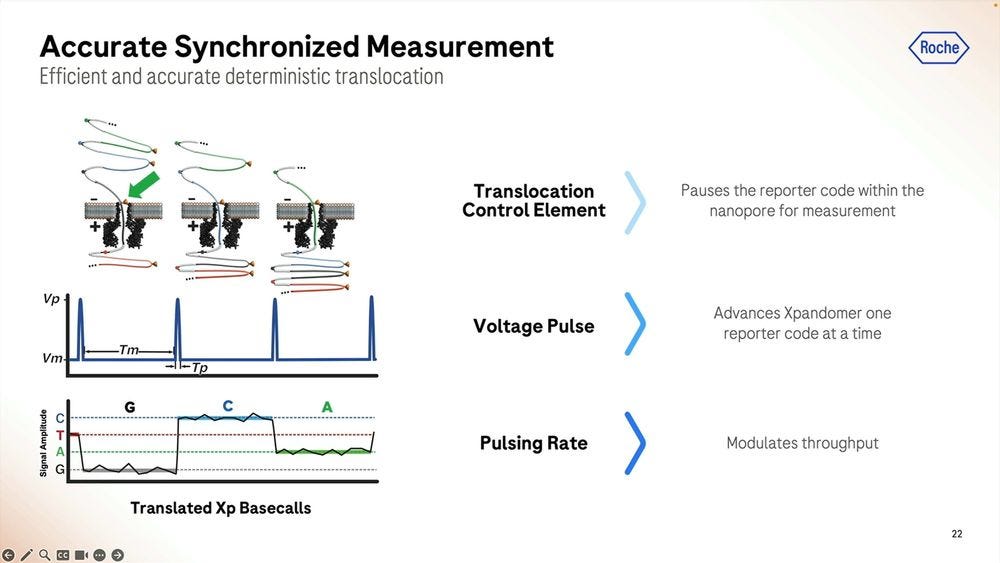
More details on how the SBX method works: the signal is very well separated during the ‘pausing’ in between each base, here showing a G to C to A signal.
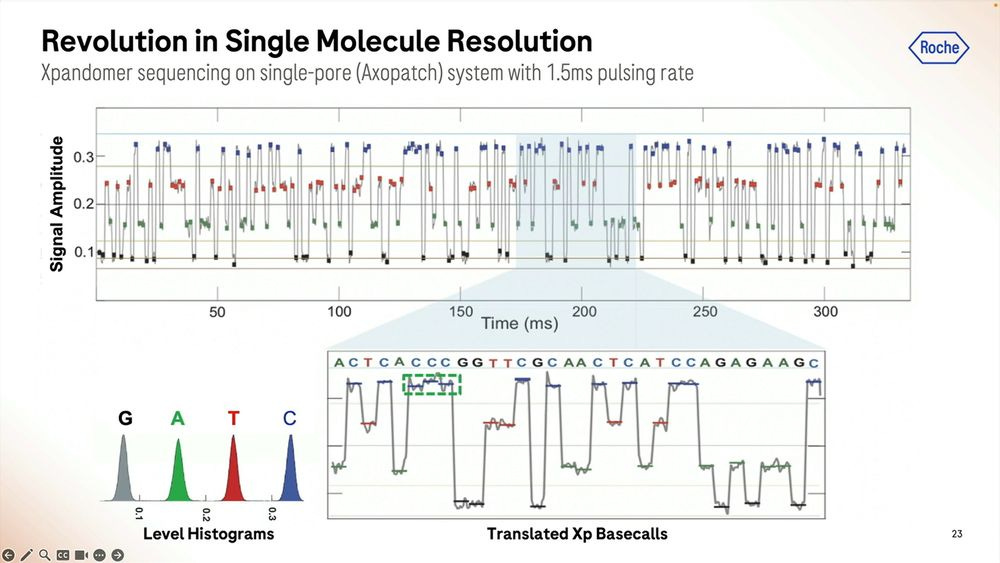
An example of the homopolymer bases, and how they look, which highlights a triple CCC. Later on they show results of how much of a problem these long homopolymers are, which seems to be only when they are longer than 15-20mers.
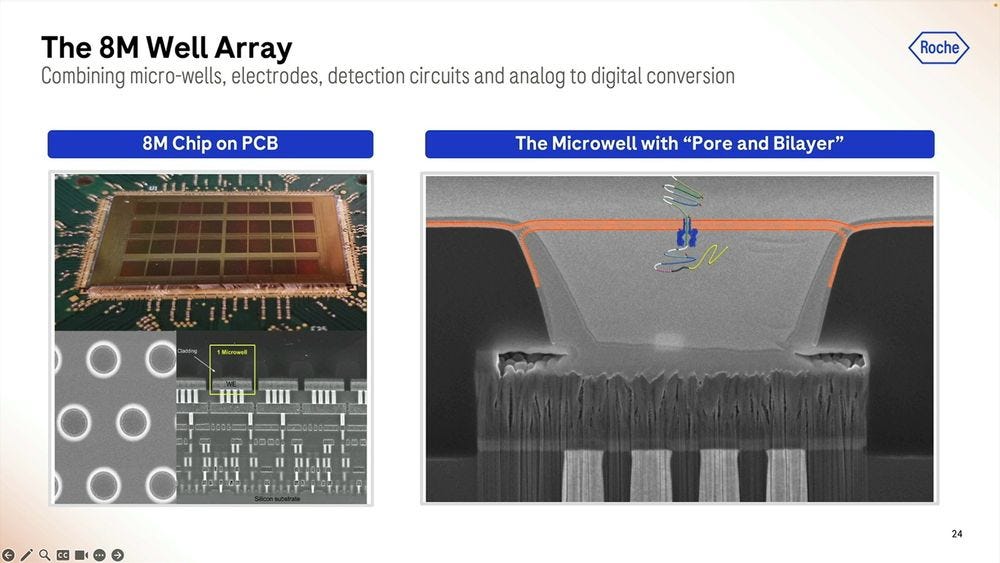
On the silicon side of things, the Genia acquisition is what brought the 8 million well chip, with the electrodes and detection circuits underneath and where the analog to digital conversion takes place. The pore bilayer is constituted “freshly” at the beginning of the sequencing run, which is one big difference to the way Oxford Nanopore does it. Instead of selling flowcells “pre-loaded” with the pores which have an expiration date, what Roche SBX does is to put the “pure silicon” chip on the sequencer machine, flow the biochemical parts to constitute the pore bilayer, and then flow the library prepped molecules to start the sequencing. This means that with an efficient “washing” of the chip, they can reuse the 8M well chip several times. The problem with this approach is that you need a large complex instrument to perform these steps, unlike the Oxford Nanopore “pre-loaded” flowcells which come in pouches the size of a chocolate bar.
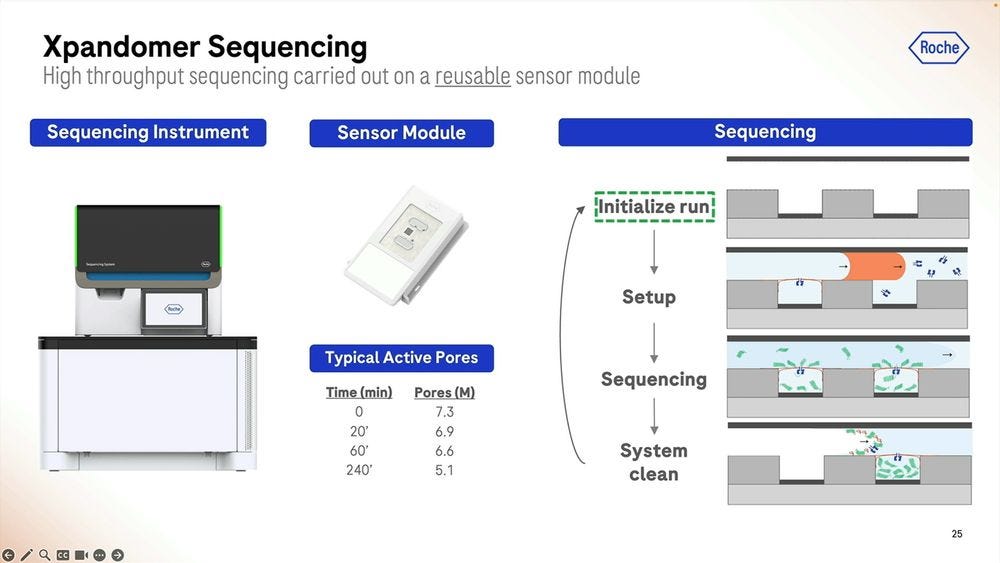
The flowing of the bilayer is done when the run is initialized, and they see a good window of operability for reusing the sensor module (the chip). Again, Illumina should be scared of this model, as it breaks what is Illumina’s model of expensive photolitographically-edged flowcells that are “one and done” and hence expensive. On the other hand, Element Bio never moved away from glass flowcells, pretty much a technology that’s been used since the eighteen century and thus very very cheap to produce and handle.
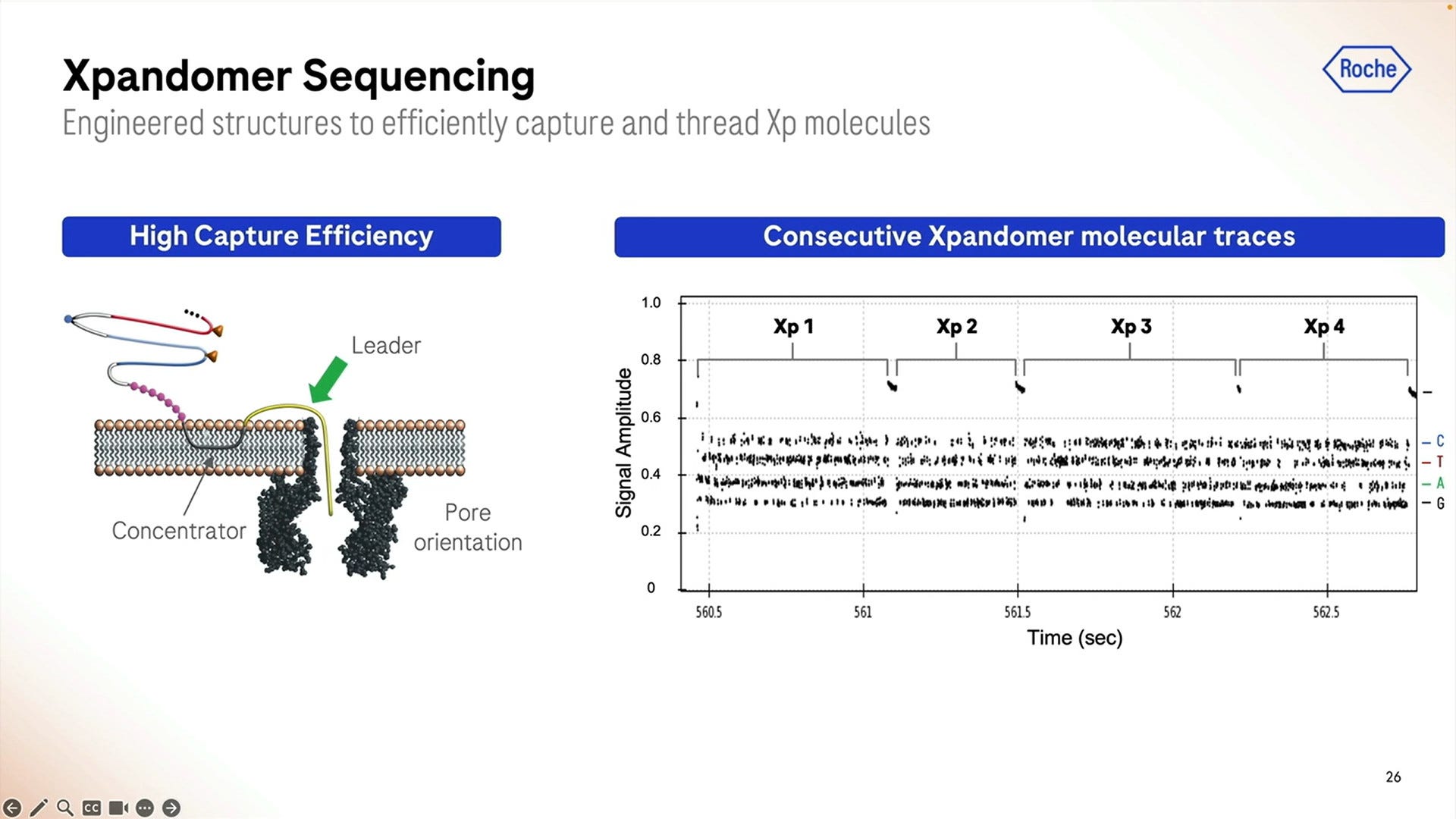
Another important detail for the SBX system to work is how to “attract” the expandomers to the pore, here they show the use of a “concentrator” which uses differences in behaviour to the leader sequence when it comes to either be attracted to be “in membrane” or to poke out of the membrane. Once they’ve been swimming in the bilayer for a while, they find a pore and the leader starts the sequencing process.
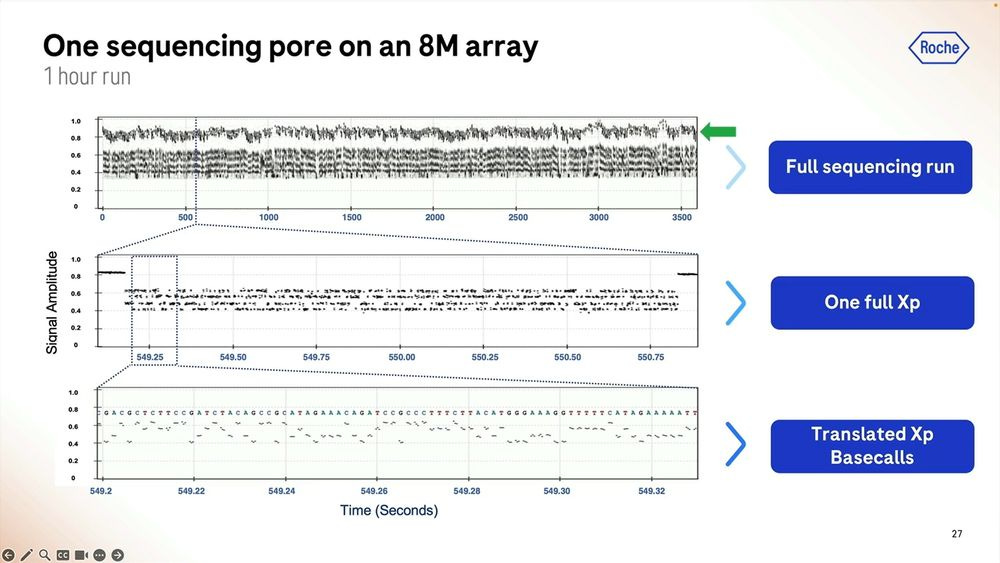
The raw signal from these is highlighted here for a short 1 hour run, and we can zoom in to each individual base called which in this case shows what happens between second 549.2 and 549.32, so 0.120 milliseconds here producing a bit less than 100 bps.
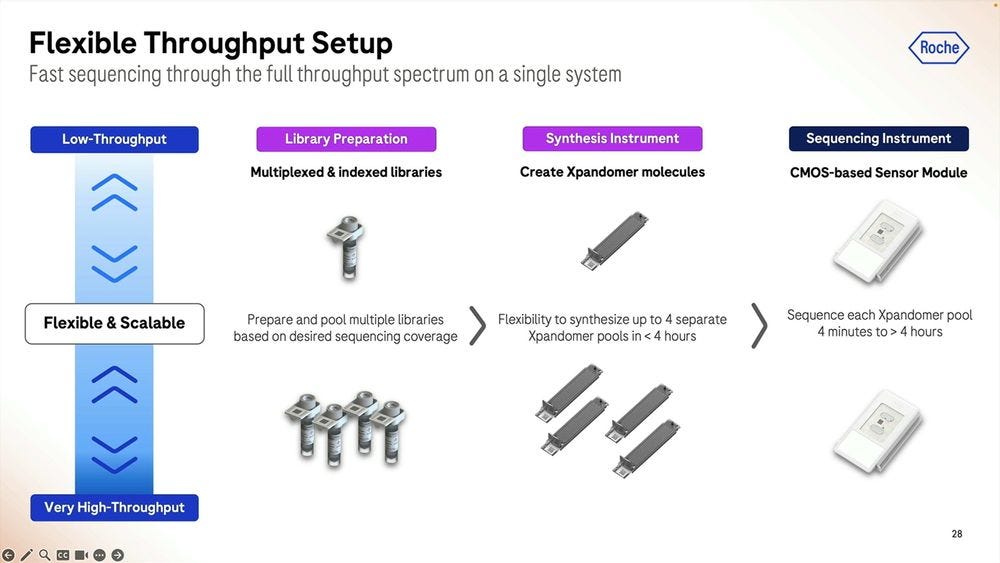
The SBX system is designed for flexibility, from multiplexed and indexed libraries that can be pooled into the synthesis process, with up to 4 expandomer pools going into the instrument which can run anywhere between 4 minutes to 4 hours.
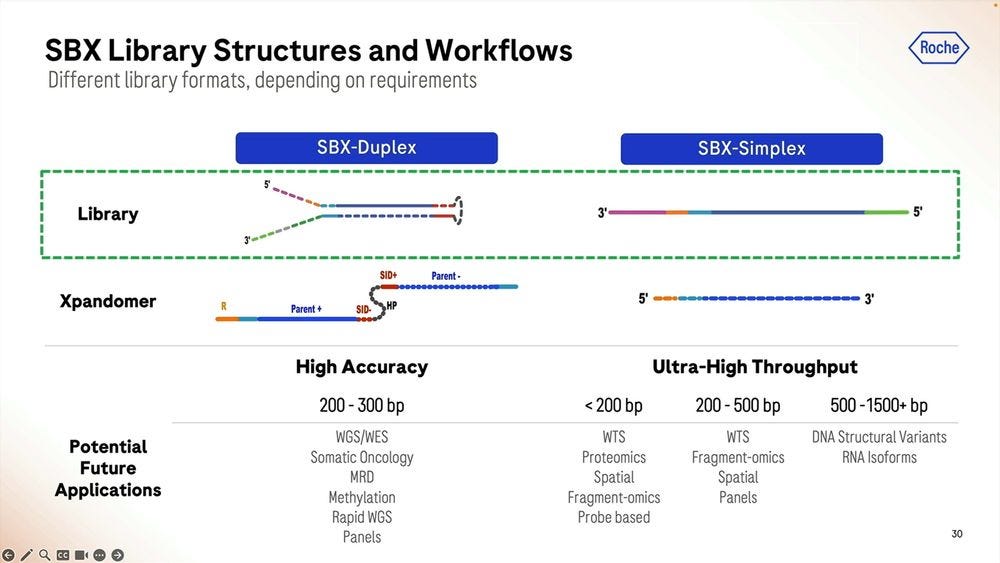
So given that there are two types of readouts, Simplex and Duplex, how is the Duplex library prep done? It’s similar to other methods used in NGS, where the double-stranded DNA is “hairpinned” and the single-strand molecule is stretched out and the adapters added. This means that the Duplex high accuracy readouts will be roughly have the length as the Simplex equivalent, in the 200-300bp range, to cov3er WGS/WES applications, Somatic Oncology, MRD, Methylation, Rapid WGS, and cancer and other panels.
The Simplex method is in comparison higher throughput but lower accuracy, and here it depends on how long the starting molecule is, similar to what happens in Illumina sequencers. One can have molecules shorter than 200bps, and many of them, for transcriptomic counting (WTS), Proteomics counting, Spatial-omics, Fragment-omics for Liquid Biopsy diagnosis, anything probe based. At the 200-500bp range, the Roche SBX will produce fewer reads, and even fewer at the 500-1500+ bp range, where these lower accuracy Simplex reads can be used for applications like DNA Structural Variants or RNA isoforms.
This is probably the most important point in the presentation in terms of the “hidden numbers”: the longer the molecule, the lower the count of Simplex reads the machine will produce. And the longer the hairpinned molecule, the more inefficient the Duplex process will be, so currently Duplex is only useful for the 200-300bp range.
This means that Roche SBX sequencing is a high-throughput short-read sequencer, with many similarities to Illumina and Ultima Genomics. Although it can produce long-ish reads, these are going to be at low throughput, which means that the cost per read will be higher, and they will be Q20 accuracy, which means that then they’ll have a tough time competing with Oxford Nanopore sequencing equivalents. PacBio doesn’t do 100M’s of long reads, unless with library prep tricks that can only been demonstrated for narrowly defined transcriptomics, but it does compete with Roche SBX in applications like Structural Variation.
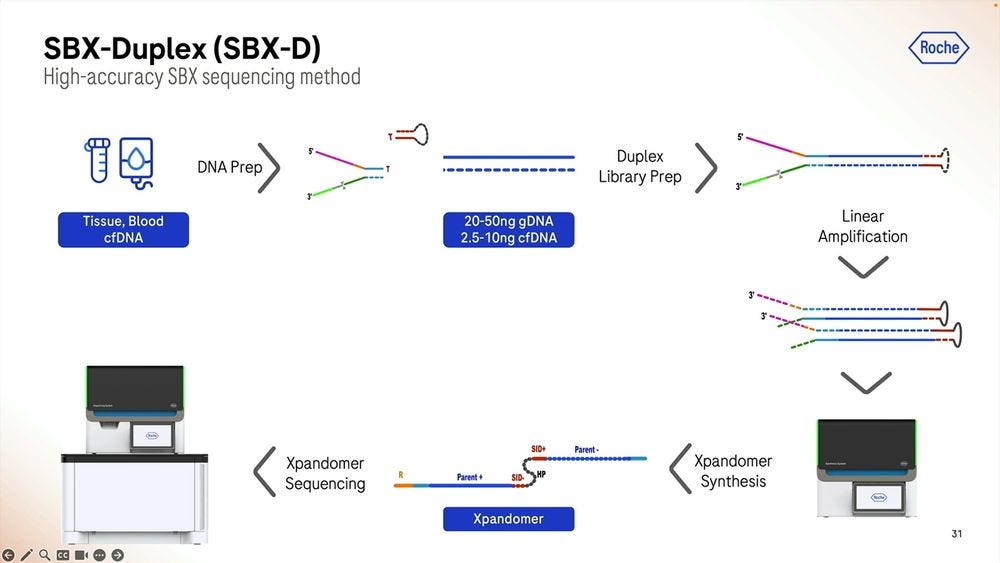
The SBX-Duplex method can operate on the 20-50ng genomic DNA range or at the 2.5-10ng cell-free DNA range. Putting this in context, a 10-20ml draw of blood, the size of a lipstick, will deliver 6-12ng of cell-free DNA, depending on how well extracted from the plasma after spinning down the blood cells.
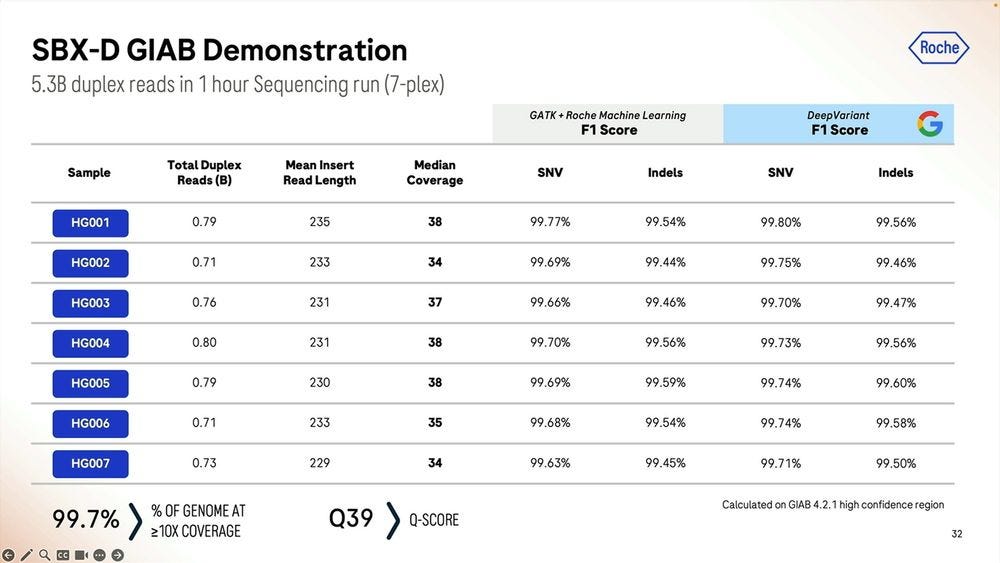
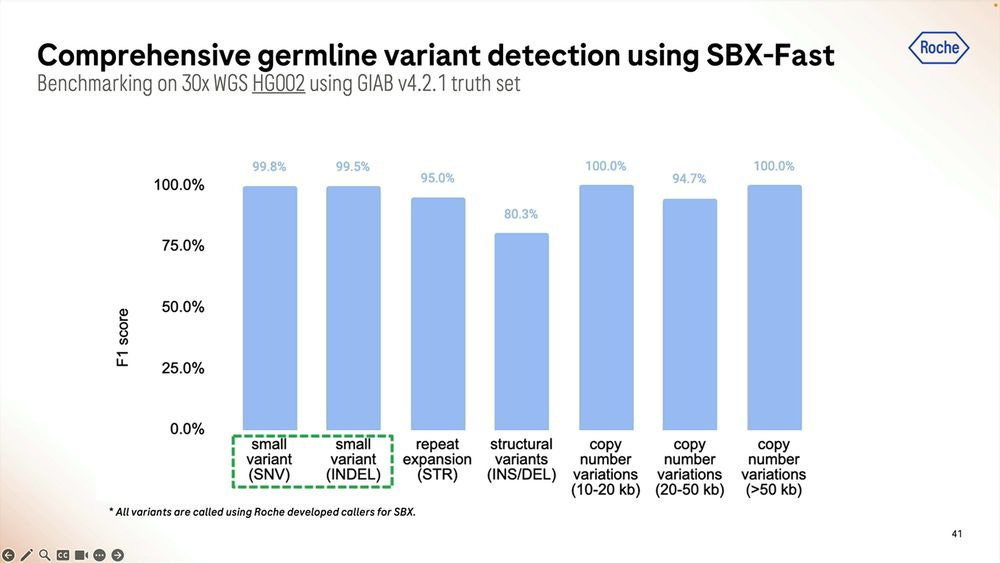
Given a cohort of genomes that have been well studied, the Genome-in-a-bottle HG001-HG007 cohort, the SBX-Duplex (SBX-D) methodology can produce 5.3B duplex reads in a 1 hour sequencing run, or roughly 0.7-0.8B reads per sample, with median coverage of 34-38x. Analyzing the mapped data with GATK plus Roche’s ML method, it produces F1 Scores of 99.60-99.80% for SNVs and 99.40-99.60% for Indels. The Roche SBX data has also been trained on Google’s DeepVariant software, which achieves roughly another 0.02-0.05% F1 score percentual points in SNVs and 0.02-0.10% F1 score percentual points in Indels.
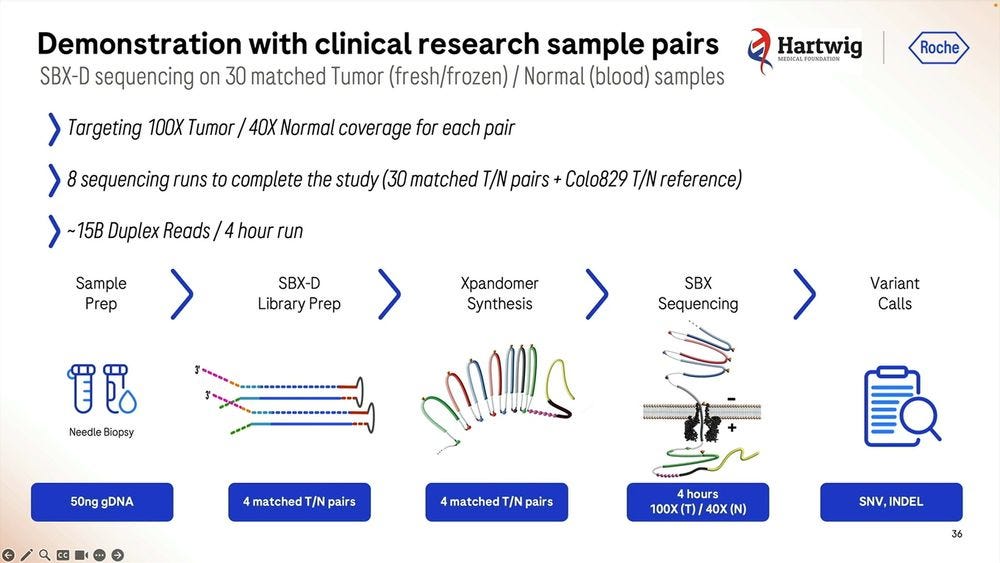
They gave this Roche SBX technology to Hartwig to replicate in beta testing, the machines were operational the day after unboxing, and they produced a tumor-normal pair of samples (100X T / 40X N) for somatic variant calling. They did 8 sequencing runs for 30 matched TN pairs plus the Colo829 TN reference. The runs were done with 4 hour configuration, delivering 15B Duplex Reads for each 4hr run.
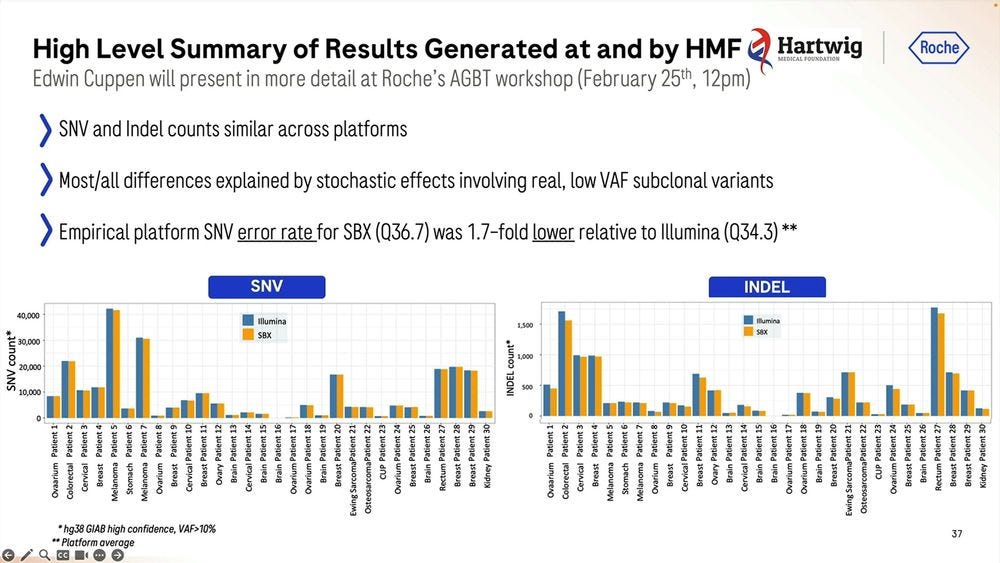
The empirical platform somatic SNV error rate was 1.8-fold lower relative to the Illumina equivalent, which again, should be a concern for the Illumina fans.
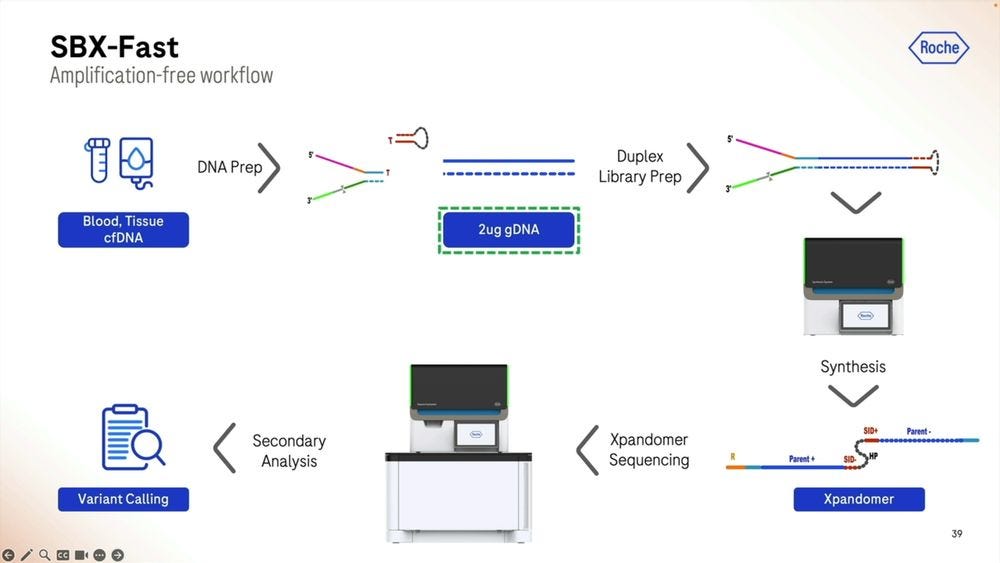
Another way to operate this system is in SBX-Fast mode: here we sacrifice input DNA amount to have a faster Turn-Around-Time (TAT). Here we are using 2 micrograms of DNA, equivalent to 2000 nanograms, which would mean bleeding out a a patient to death if we were trying to extract cfDNA, but for other cell-based samples, it’s no biggie.
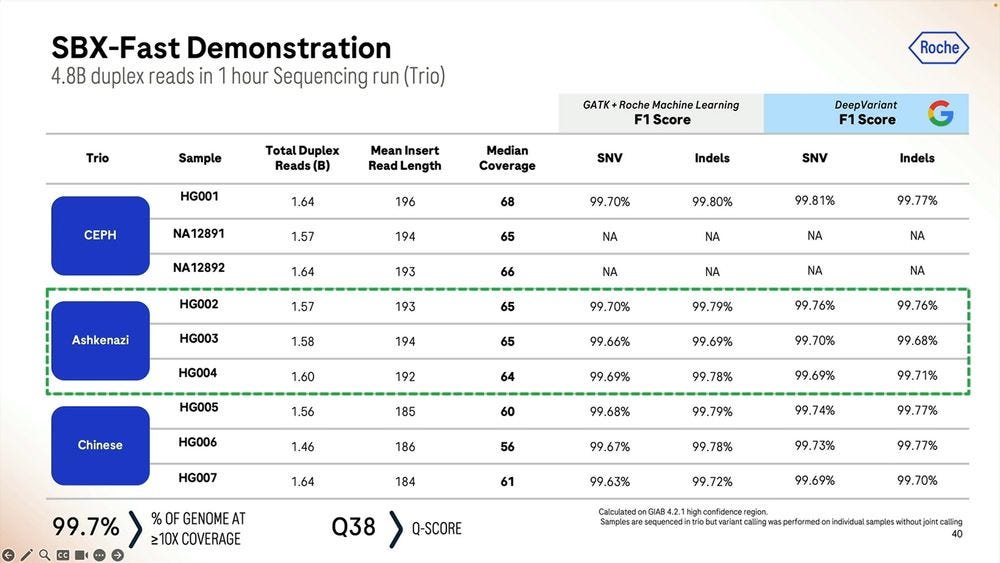
One can do shorter sequencing run times of 1 hour, which deliver 4.8B duplex reads instead of the 15B equivalent in a 4 hour run. So the system degrades in performance after the first hour, because it produces about 5B reads per hour in the first hour, but only 3.75B reads per hour in a 4 hour run. This is the same as to say that a 4 hour run is only 75% efficient per unit of time compared to the 1 hour run.
The metrics for SBX-Fast, not dissimilar to what is shown for the 4 hour runs.
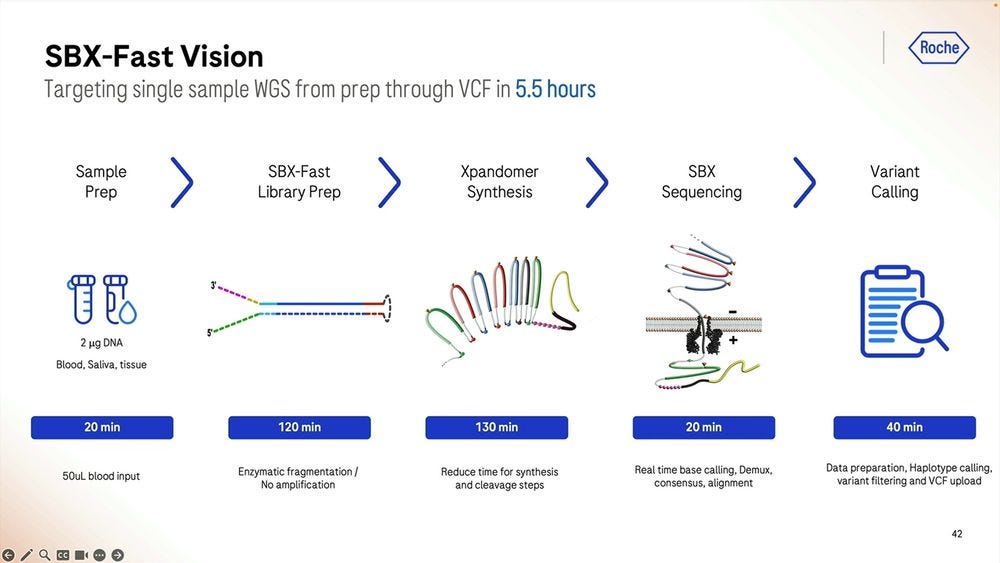
So the SBX-Fast vision taken to the extreme is that one can go from blood/saliva/tissue sample to VCF in 5.5 hours, with the longest process actually being the library prep, which is 2 hours plus 2 hours and 10 minutes starting with 2000 ng of DNA. Here we only do 20min of sequencing, which if we turned into 60min as the previous slides show, then it’s 6 hours and 10 minutes total.
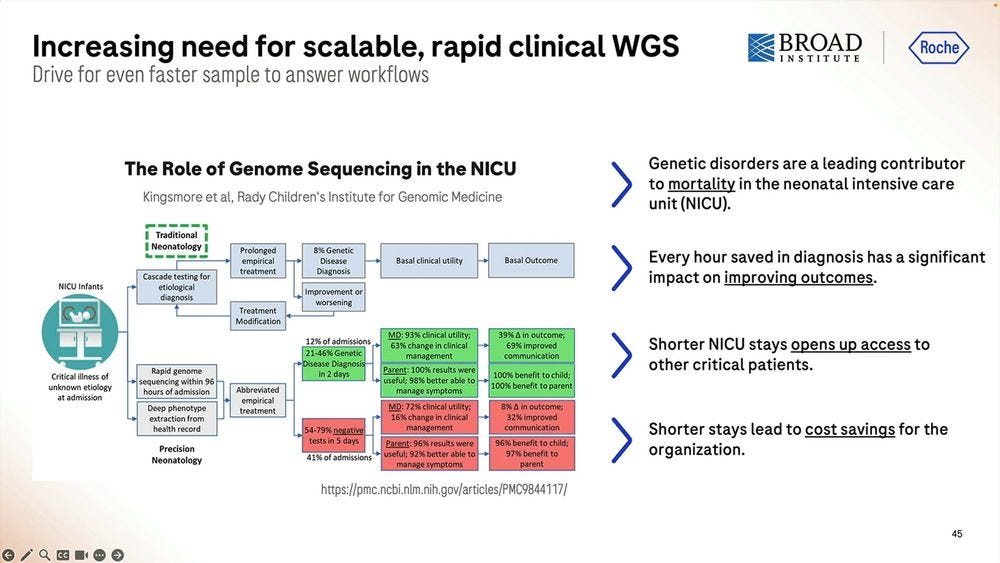
One application of this fast methodology is NIPT/NICU. Examples from the Broad Institute and the Kingsmore lab at Rady Children’s Institute are a-plenty.
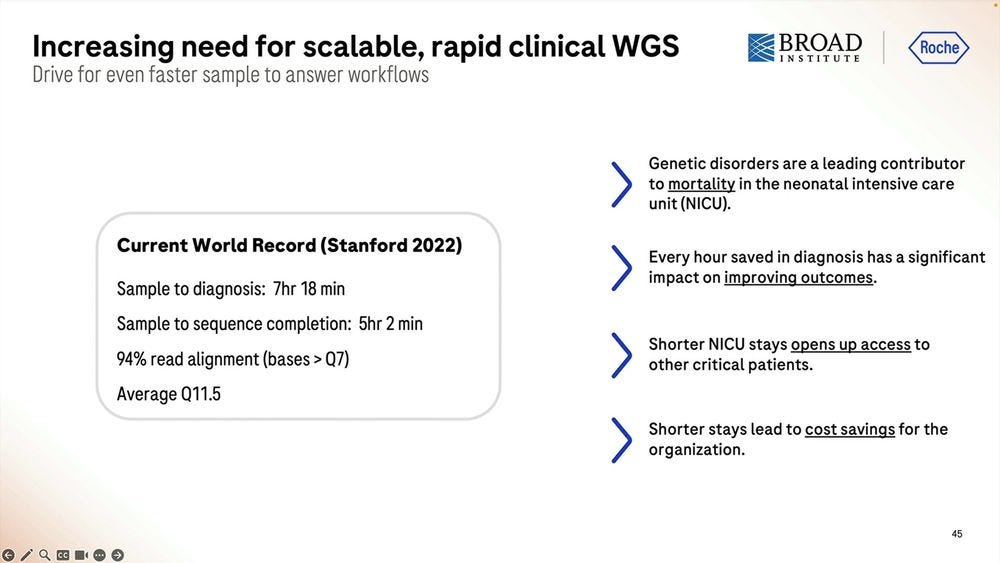
The alleged current record with Illumina sequencing is 7hr 18 min, so Roche SBX-Fast here is either 5.5 hours with not a lot of throughput or 6.2 hours with a 1 hour 4.9-5B read throughput.
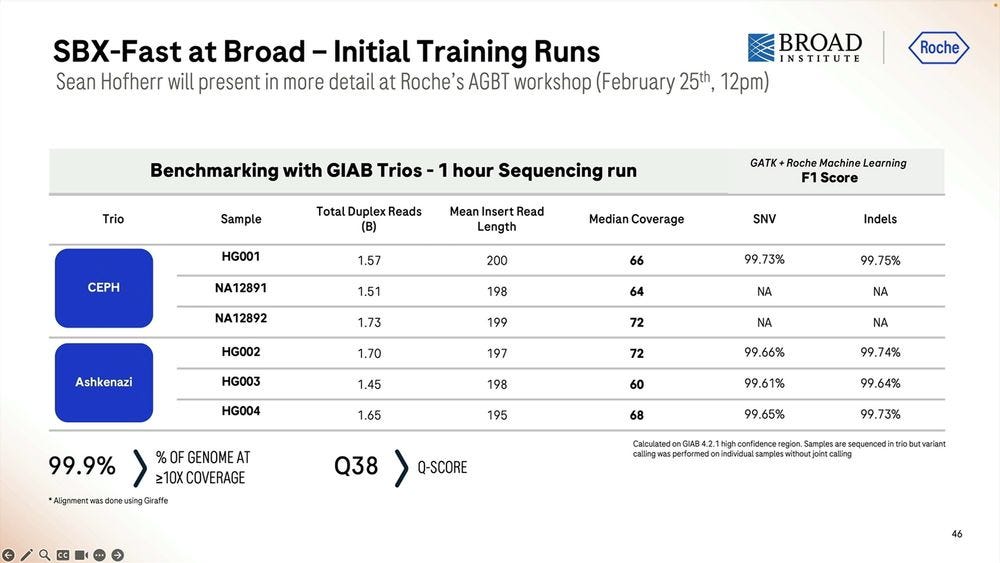
The numbers shown above are for 1.5-1.7B reads for 20 min runs with mean insert length of 195-200bp, which is pretty much 1/3 of the throughput of the 60 min run. So the system degrades in performance “after the first hour” reaching 75% efficiency at the 4 hour mark.

More numbers for Coriell samples, anybody can buy these and test them on their favorite NGS method to compare the performance.
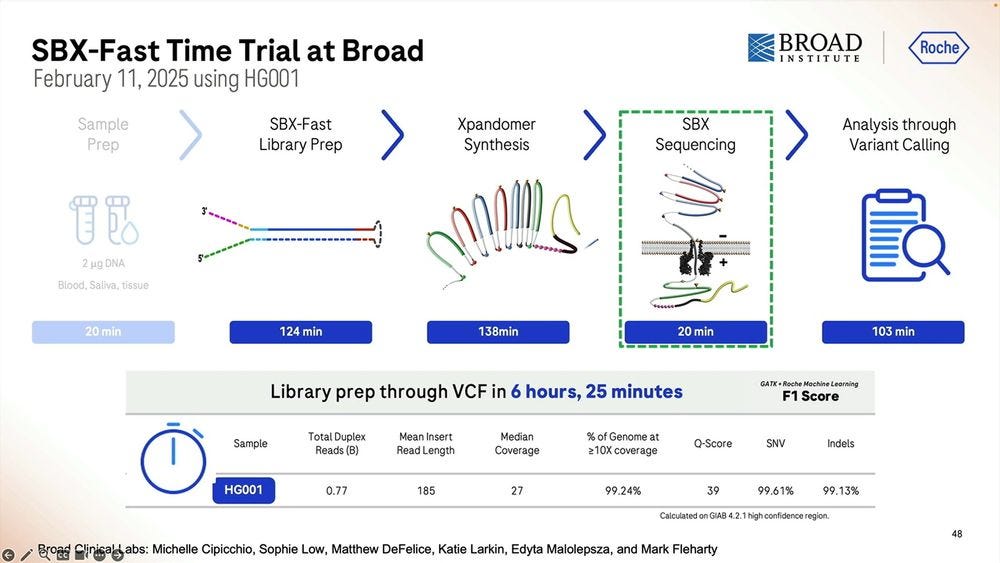
The trial of SBX-Fast at the Broad took longer than 5.5 hours, here 6 hours and 25 minutes in the capable hands of Broad’s employees.

The Duplex technology was explained in more detail by John, while Mark went back to sit on the couches behind and have a sip of coffee.

The specs of the machine repeated: 500M bases per second of raw data, or 15B duplex reads in 4 hour runs, with an mean insert size of 200bp.
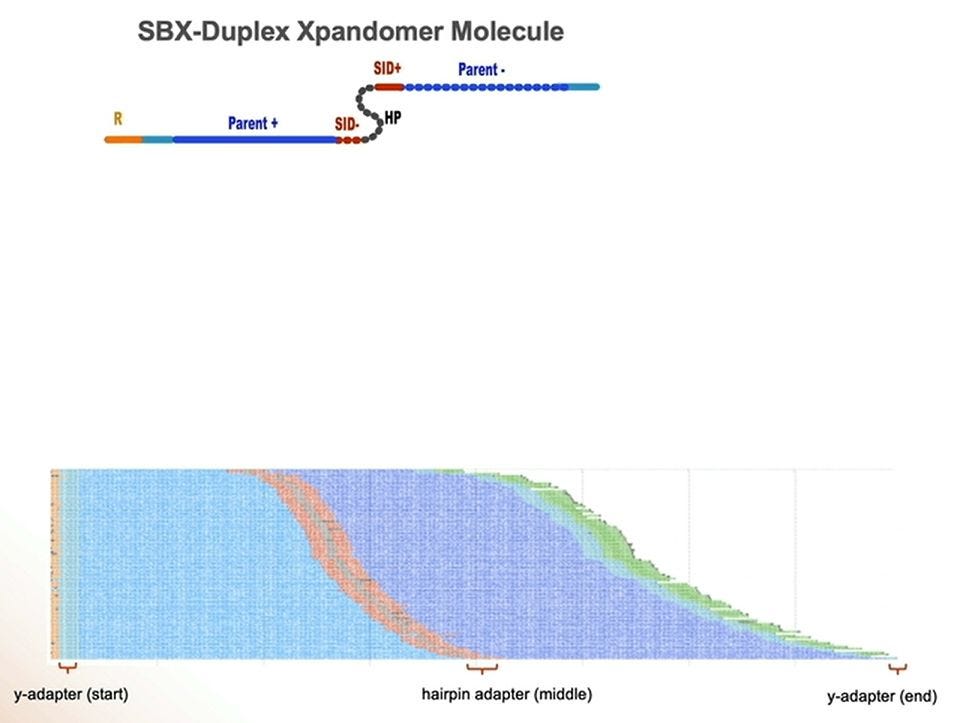
A pile of duplex reads, with the hairpin adapter in orange in the middle.
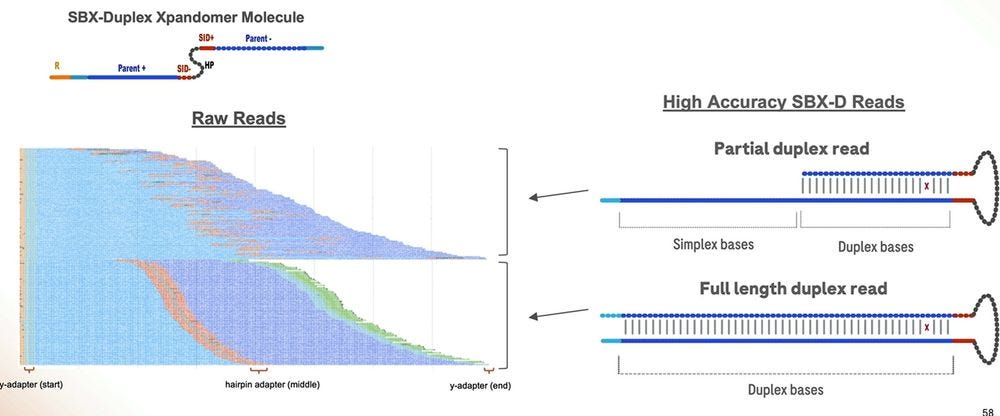
The machine also produces partial duplex reads, which are useful, but aren’t high accuracy end-to-end.
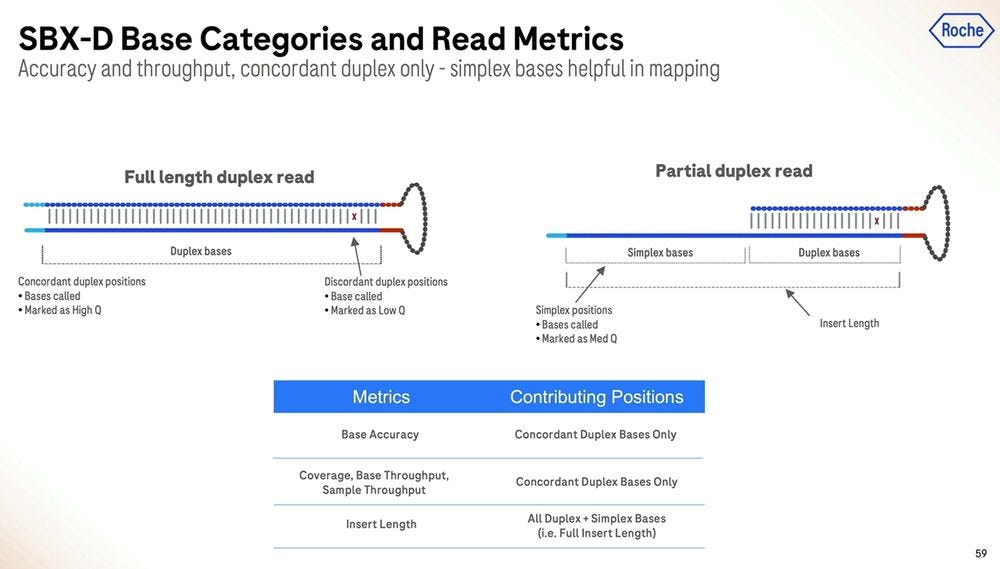
The partial duplex reads are concordantly high accuracy for the part where they are duplex, and equivalent accuracy for the part they are simplex, which means they can be equated to either type in their composition, and all of this can be reflected in the FASTQ/BAM formats everyone has been using in the last couple of decades.
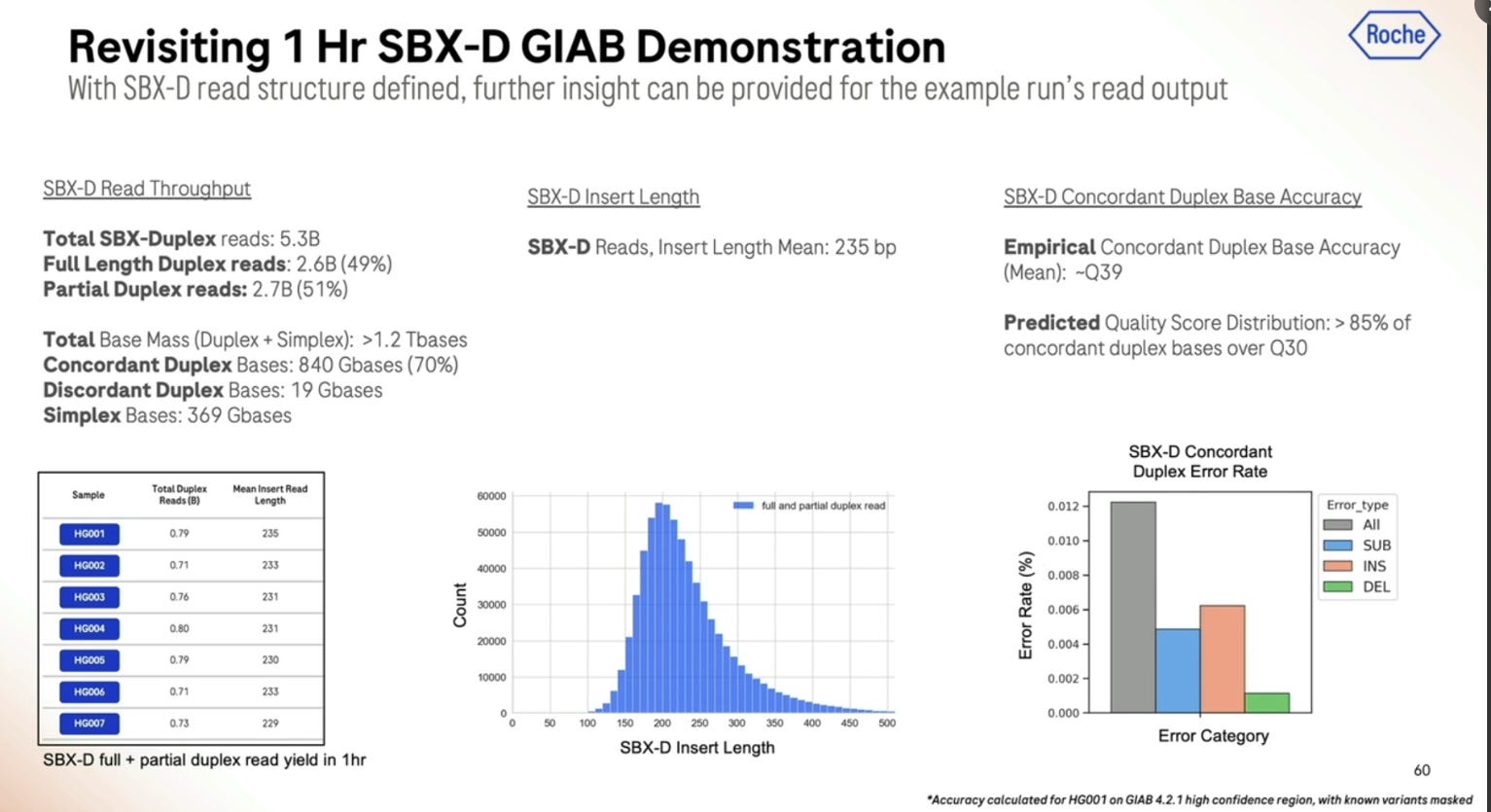
Here is another important slide: only about 50% of the reads in a 1 hour run are fully SBX-Duplex, with the other 50% being Partial Duplex reads. These 5.3B SBX-D reads produce 840 GB of concordant duplex bases, which gives me an insert size of 168bp if they were all the same length. So the 200bp is more of a modal insert length, with the longer fatter tail being on the “good side”, but dropping precipitously at the 300bp mark. So it is true that SBX-D is 200-300bp, but it’s more 200bp modal or 235bp mean length with a small percentage at the 300bp range.
Another important metric is the per-read accuracy, which Roche quotes at ~Q39 mean quality for duplex bases. This sounds high, but then the distribution goes to >85% over Q30 when looking at the total mass of bases, with insertions and deletions being more predominant than substitutions. So to summarize, it’s >85% above Q30, more micro-indels than substitutions, which is not what’s seen in Illumina SBS (ratio more like 1/10) but similar to what’s seen in Ultima Genomics reads.
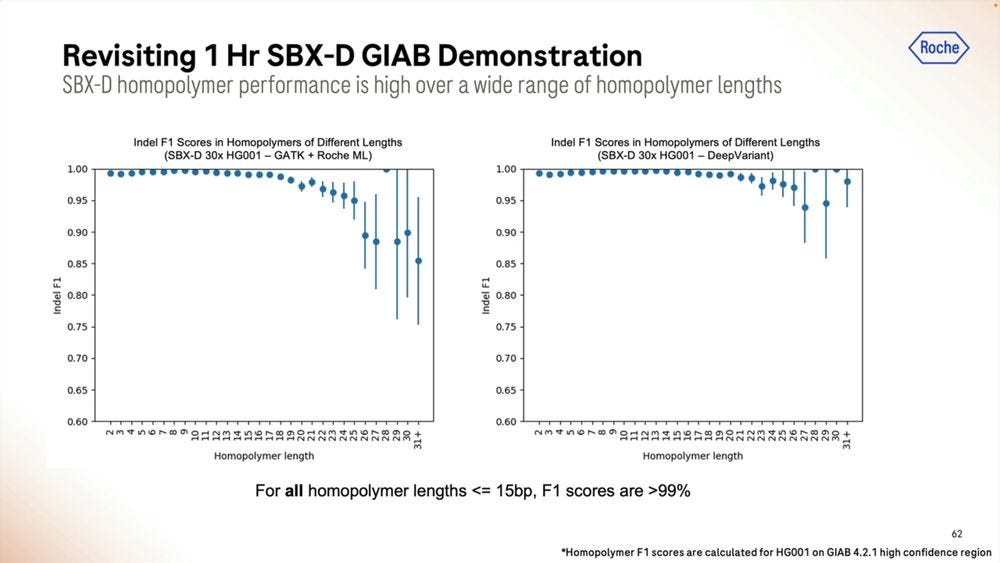
The homopolymers start being a problem for mapping applications in the 15-20mer range. The algorithms matter here, with DeepVariant dealing better with them than GATK + Roche ML software.
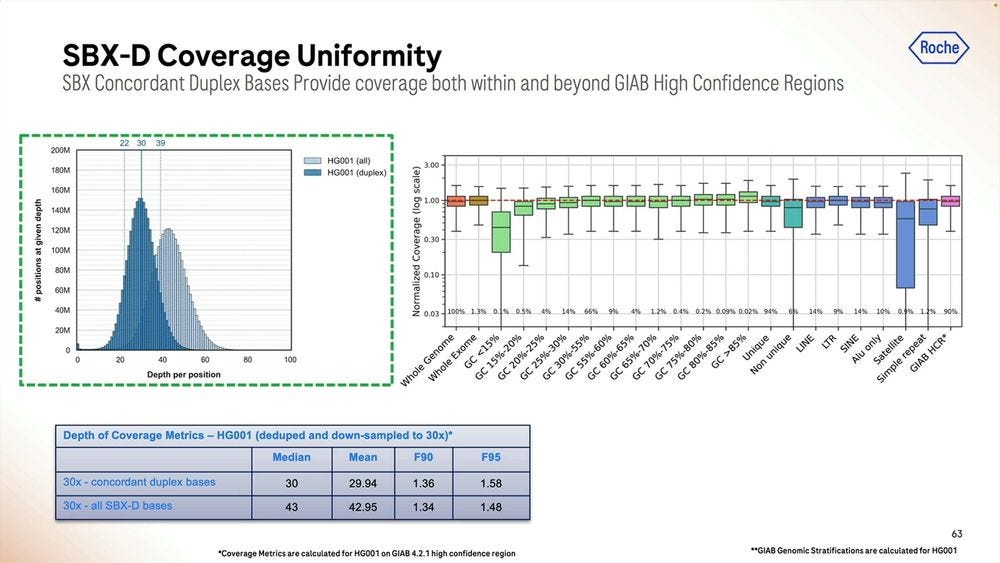
The coverage uniformity is decent, and the replicability from sample-to-sample is also good. The GC bias is not bad when mapped to the human genome, comparable to similar sized reads done with Illumina SBS.

How to handle the Primary Data Analysis when dealing with this Ultra-high throughput NGS instruments? Roche sees a combination of cloud and local as potential solutions. Local means there is the need for a 3rd machine, the local compute.
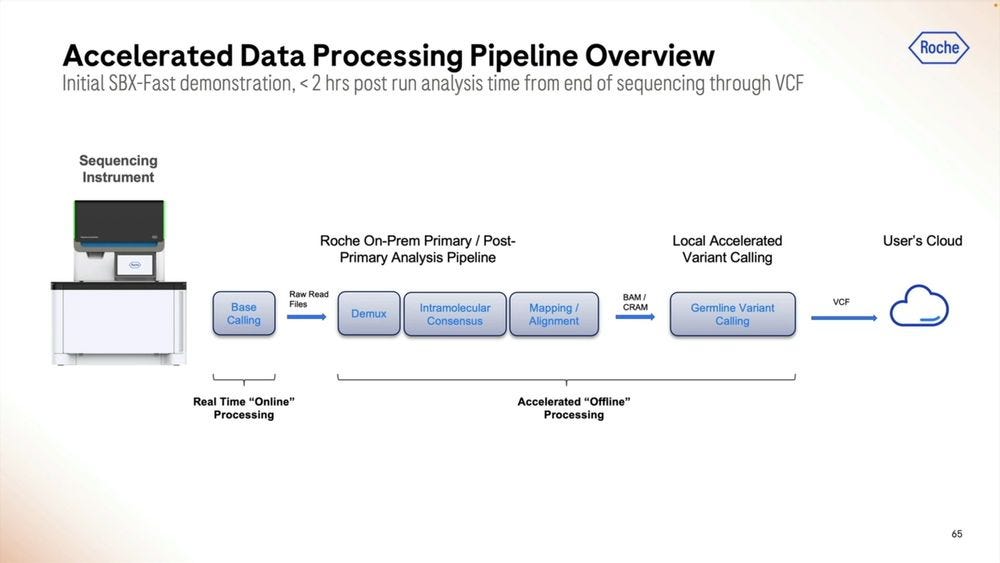
Given the right algorithms and parameter sets, the post run analysis time can be squeezed into less than 2 hours, here with Real Time “online” processing of the signal into FASTQ reads.
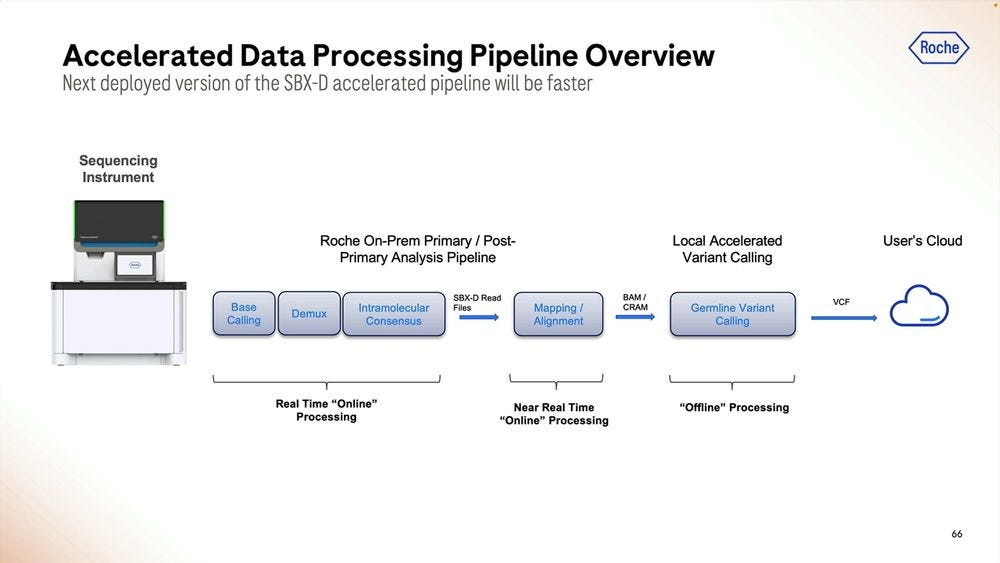
The accelerated version would incorporate more of the primary analysis steps into the Local compute. This is all highly paralellizable.

We now switch gears into other applications beyond human WGS/WES done with SBX-D of 200bp inserts. Here showing two extreme cases, one doing 10X Genomics Flex Probes (168bp), which produces 13.7B reads per hour and 2.3Tbs per hour. Crucially, if the inserts are 4.6x larger, the reads per hour metric drops to 1.8B, or 7.6x lower throughput. So there is a penalty associated to longer reads, and it’s a bigger penalty than simply proportional to the length.
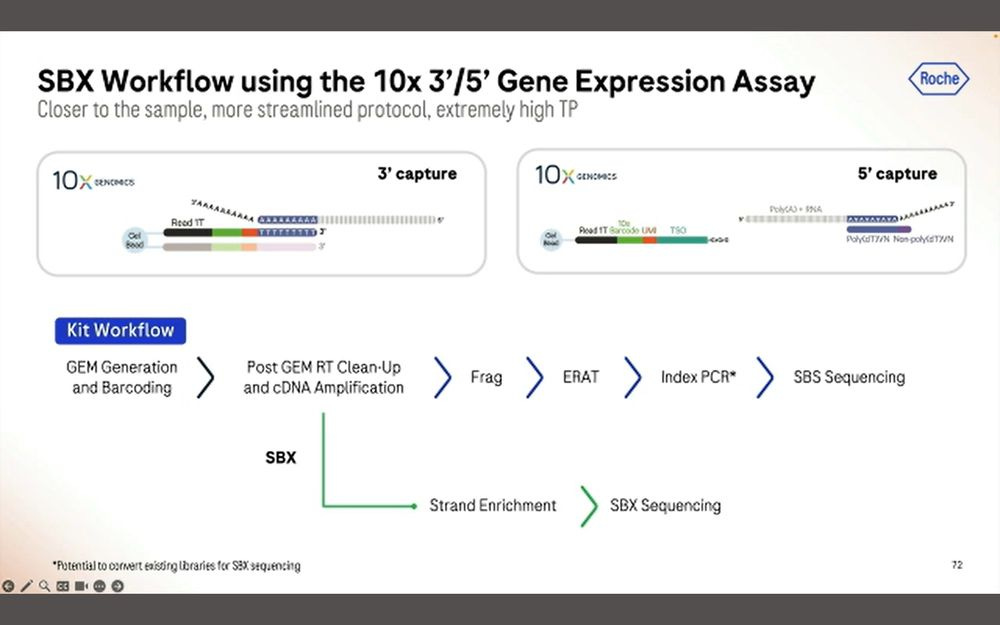
Another application is longer molecules of the kind that comes out of the omnipresent 10X Genomics Chromium instruments.

Similar to what’s been seen before by Ultima Genomics, one can artificially turn the SBX simplex reads into fake “paired-end” reads so that the Illumina-specific cellranger software runs with SBX reads.
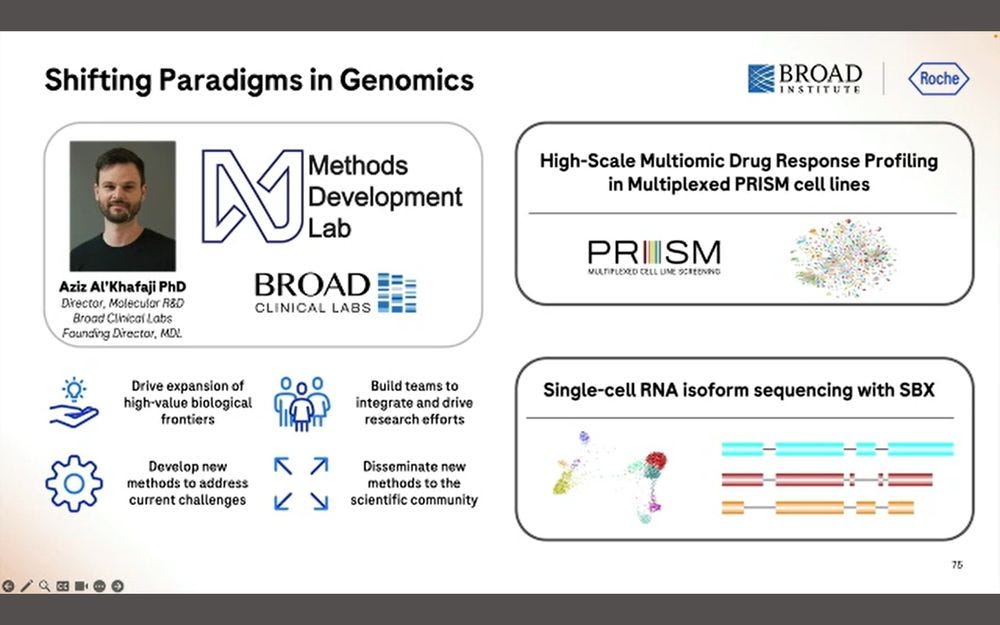
Two types of applications linked to 10X Genomics Chromium (maybe also Visium?) using SBX Simplex reads: High-Scale Multiomics Drug Response profiling, and Single-cell RNA isoform sequencing.
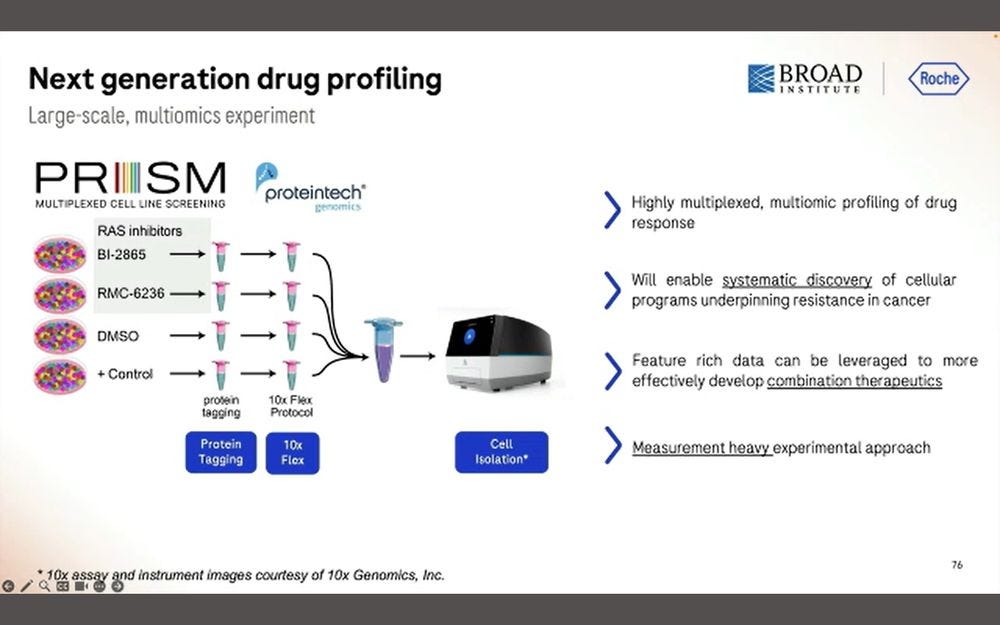
The first one benefits from the 10x Flex tagging, which is a short-read counting application.
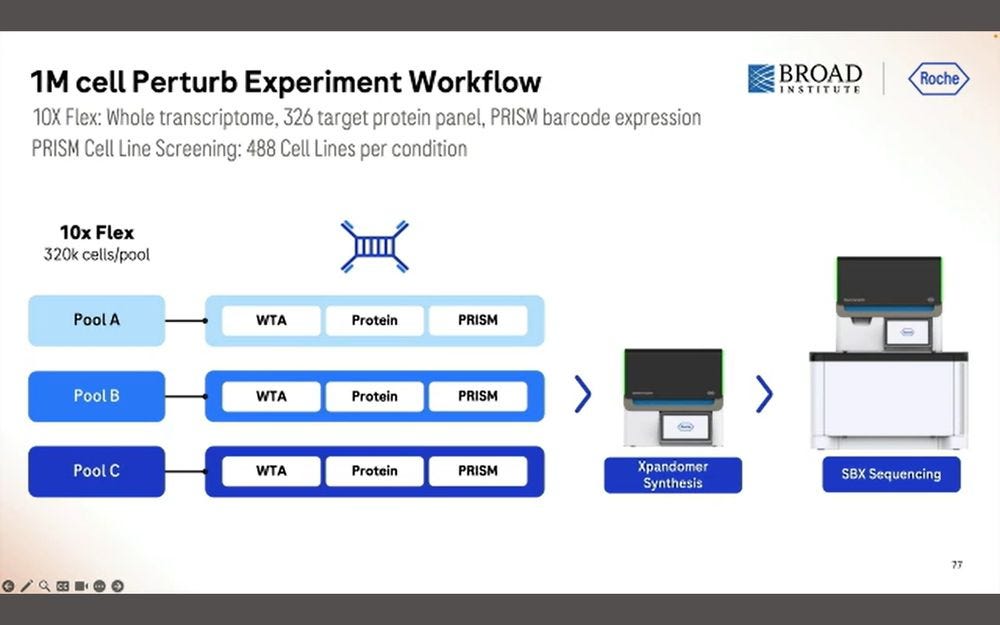
Here they put 326 target protein panels, combined with the WTA library and the PRISM library, all into the same Roche SBX process.
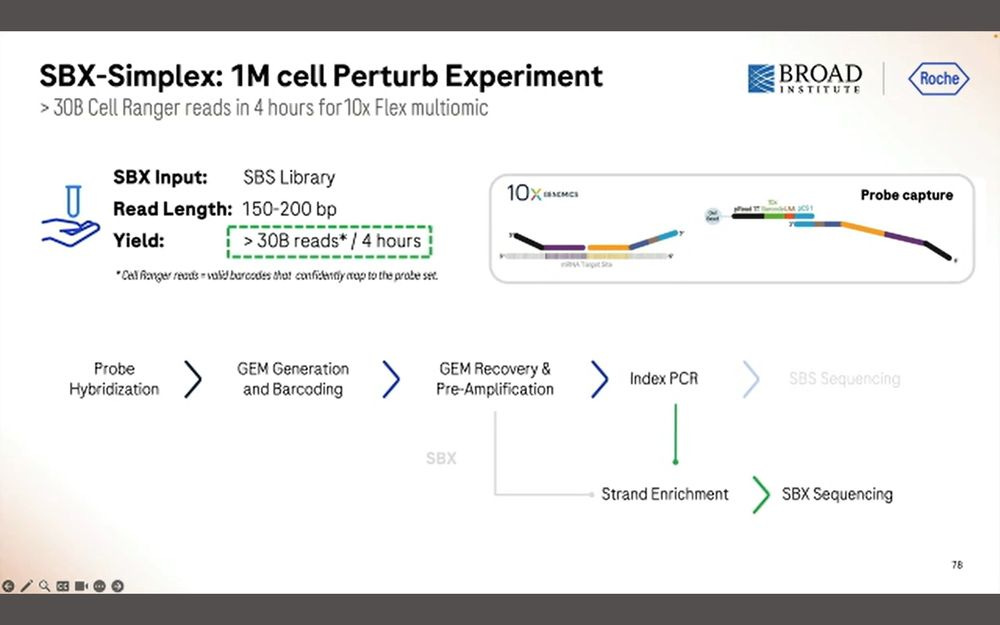
They produced 30B simplex reads in 4 hours, which equates to the 15B duplex equivalent in previous slides.

Lots of interesting science can come out of these type of methods.
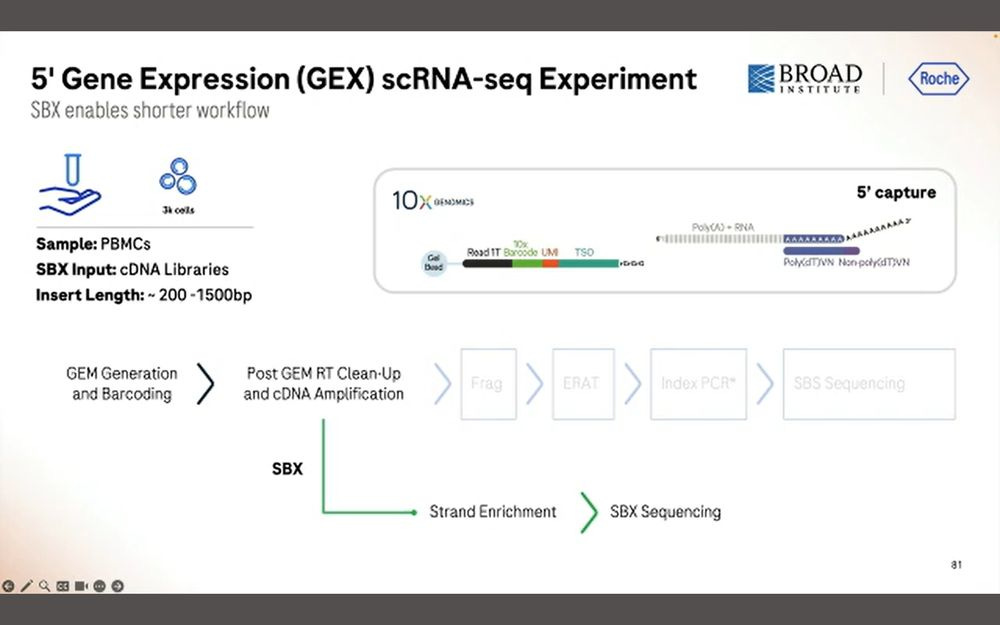
The other application is the 5’GEX method that 10X Genomics sells. Here we want to sequence the entire cDNA molecule for each of these transcripts.
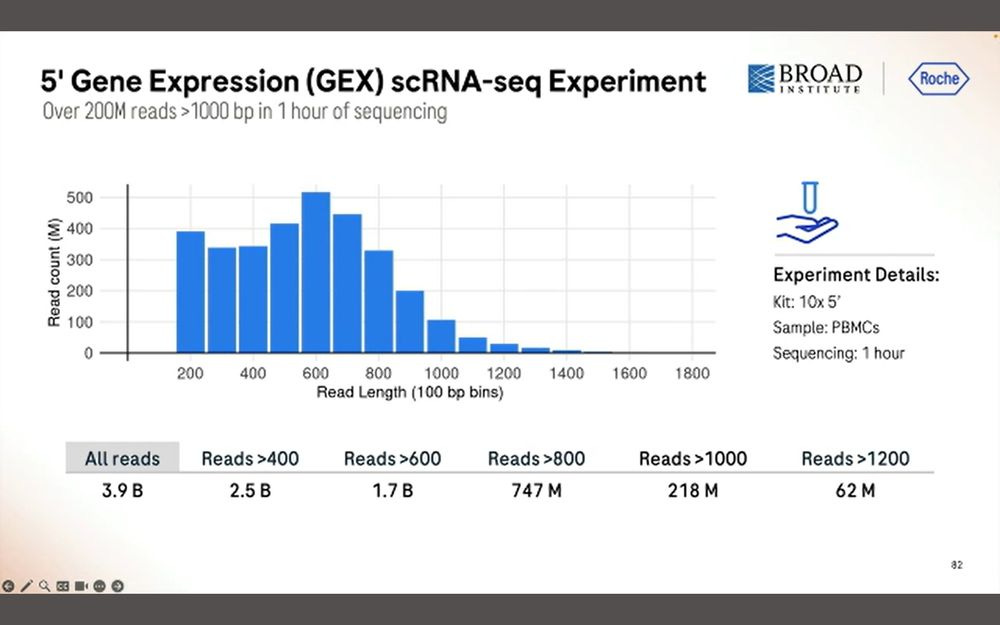
After 1 hour of sequencing, we obtain 200M simplex reads longer than 1000bp, a total of 3.9B reads with a peak at 600bp. Again, this means that there is a penalty in throughput when sequencing longer inserts.
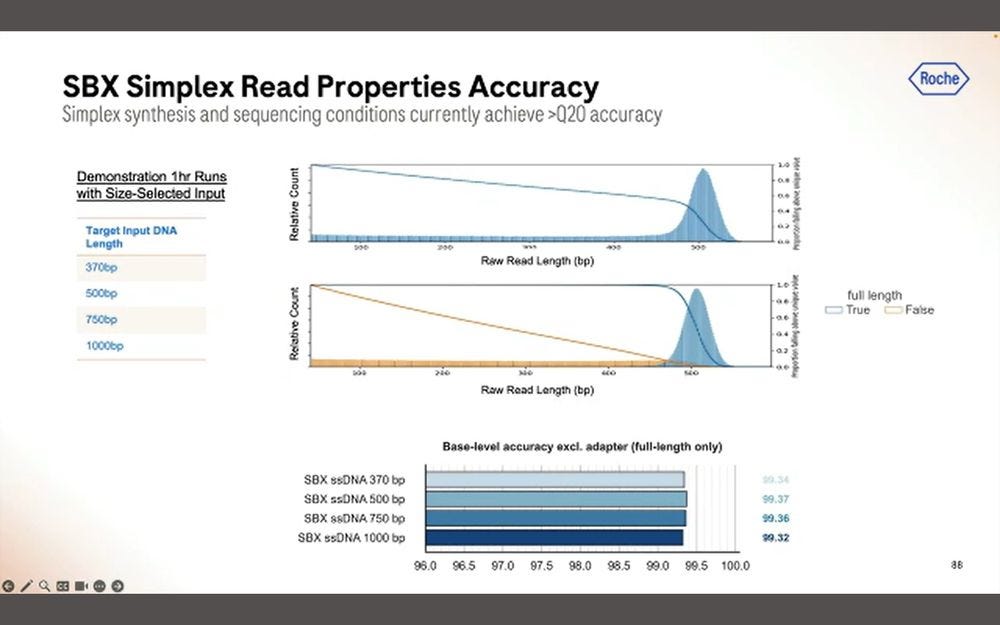
The good news for Roche is that the base-level accuracy is fairly consistent across the length of the read.
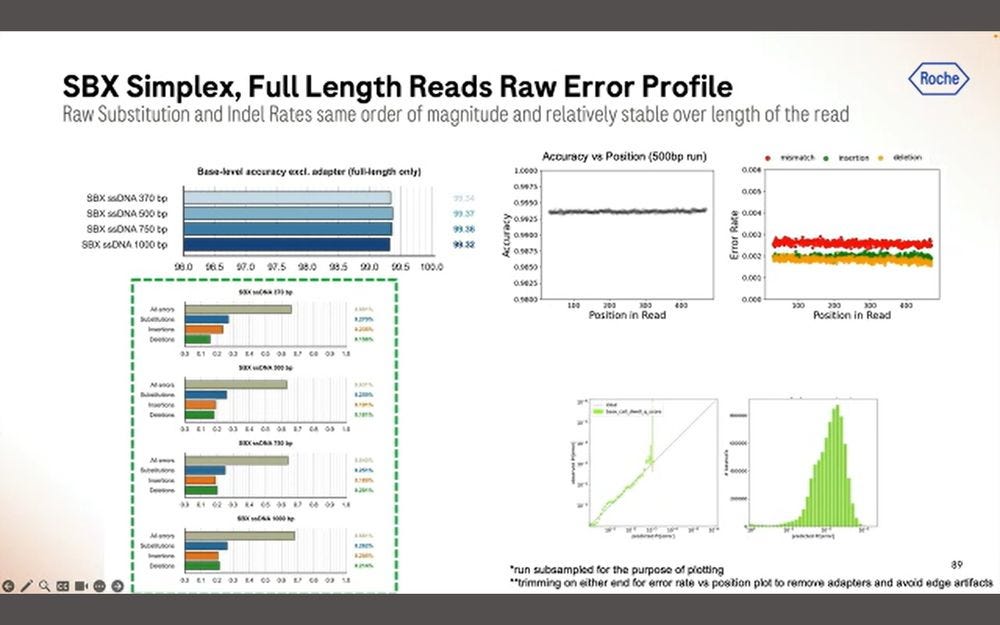
Drilling down to the type of errors, fairly similar to what was shown before: combined micro-indel error rate is bigger than mismatch error rate.
Final slide to describe what SBX will look in the future. The Reusable sensor module component is important, but too early to say how important.
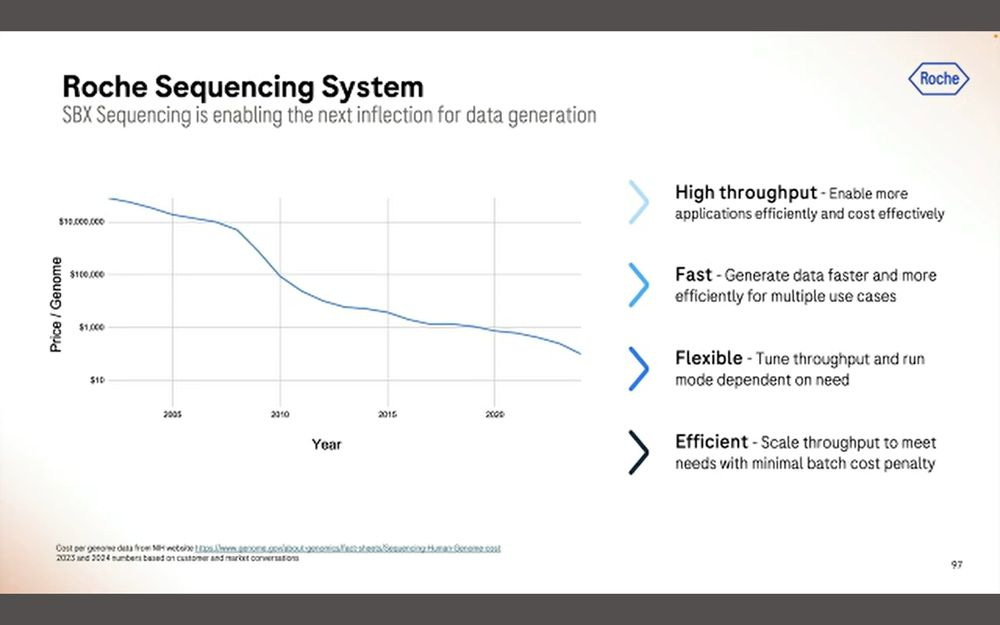
In terms of cost per gigabase ($/Gb), the presentation showed that it’s been going down over time in the NGS market, but they didn’t commit to a number for SBX-D or SBX-Simplex.
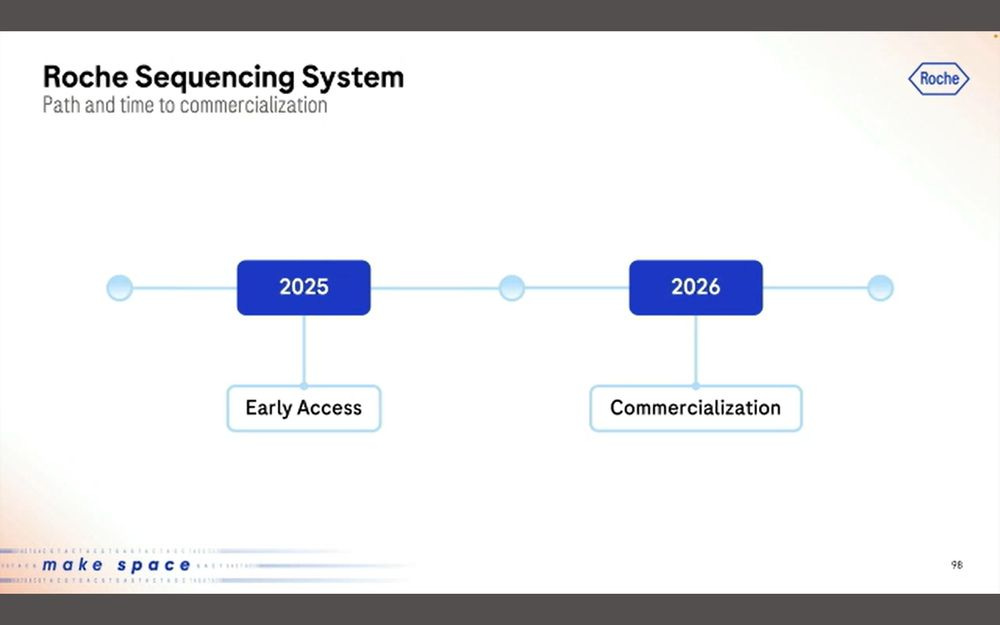
Since the Broad and Hartwig have already done Early Access, the 2025 box can be ticked off, and now the plan is for 2026 commercialization.
Based on all this content, I’ve decided to create 3 “modes” of Roche SBX sequencing. A simplex mode with a 4 hour run, delivering 12B reads of a modal 600bp length, with a Q20 modal accuracy. A SBX-D mode with a 4 hour run, delivering 15B reads with a modal 200bp length. And finally a SBX-D mode with a 1 hour run, delivering 5B reads with a modal 200bp length, both SBX-D at a Q30 modal accuracy.

This means that if a lab fits 4 runs per day out of the Roche SBX sequencer, they get similar metrics to the Illumina NovaSeqX Plus 25B Fc runs, which take 2 days. Assuming a maximum of 10 x 4 hour runs every 48 hours, then the SBX-D sequencer would produce 150B reads, or 3x the throughput of a NovaSeqX Plus. Given that the library prep is a bit longer than 4 hours, one would want to have 2 library prep instruments per each Roche sequencer instrument, although a 1 to 1 ratio would work if there is enough space between sequencing runs.
Comparing this 150B reads per 48 hour window for Roche SBX-D to Ultima Genomics, one would currently need 2-3 UG100s to keep up with a single Roche sequencer. But for very short-read counting applications, Ultima Genomics recently announced their Solaris Boost run mode, which produces 100B reads per 24 hours. This would be very comparable to the Roche 150B reads per 48 hours, or 75-90B reads per 24 hours if fully maximized. In terms of pricing, nobody beats Ultima Genomics on their $1/Gb mark, although there is rumours that the biggest Illumina customers get a discount that’s at around $1.4/Gb, which is close to Ultima’s pricepoint.
But the recently announced Ultima Solaris platform now has higher throughput out of the UG100 instrumentation, and they’ve managed to break the $100 genome mark, now at $80, or $0.8/Gb in the updated cost.
I won’t cover MGI Tech (Complete Genomics in the US) as although they also sell their MGI DNBSEQ-T7 and T20x2 platforms at $1/Gb, I doubt they’ll sell many of these outside China, and given the fact that the geopolitics since Trump’s second term are going from bad to worse, I am pretty confident that’ll still be a problem for them.
Strategic Positioning
What does this all mean for the Illumina ILMN 0.00%↑ stock and the other NGS stocks trading out there?